Article Text

Abstract
Hyperinsulinism in infancy is one of the most difficult problems to manage in contemporary paediatric endocrinology. Although the diagnosis can usually be achieved without difficulty, it presents the paediatrician with formidable day to day management problems. Despite recent advances in understanding the pathophysiology of hyperinsulinism, the neurological outcome remains poor, and there is often a choice of unsatisfactory treatments, with life long sequelae for the child and his or her family. This paper presents a state of the art overview on management derived from a consensus workshop held by the European network for research into hyperinsulinism (ENRHI). The consensus is presented as an educational aid for paediatricians and children's nurses. It offers a practical guide to management based on the most up to date knowledge. It presents a proposed management cascade and focuses on the clinical recognition of the disease, the immediate steps that should be taken to stabilise the infant during diagnostic investigations, and the principles of definitive treatment.
- hyperinsulinism
- treatment
- hypoglycaemia
- neurological damage
- cation channels
Statistics from Altmetric.com
Hyperinsulinism is the most common cause of persistent or recurrent hypoglycaemia in infancy.1 It is a major cause of neurological damage and life long handicap, and the fact that the incidence of such damage (in up to 20% of survivors) has changed little during the past 20 years2 ,3 reflects the major difficulties in managing the disease.
Until recently, the pathogenesis of hyperinsulinism remained an enigma,4 but during the past five years there has been an explosion of knowledge concerning the molecular genetics and the membrane physiology of the condition, which has given not only new insights into its cause,5 but has also opened up new concepts of treatment.6 ,7
It is now clear that the uncontrolled release of insulin is the final manifestation of a number of different processes that either alter intracellular biochemical pathways of the pancreatic β cell (thereby generating abnormal signals for the secretion of insulin8), or alter the transport of cations across the cell membrane. These abnormalities perturb the stimulus–secretion coupling mechanisms that normally ensure that the amount of insulin secreted is directly related to the ambient blood glucose concentration.8
Therefore, hyperinsulinism is characterised by the presence of insulin concentrations that are inappropriately high for the concentration of blood glucose. A “normal” insulin concentration for normoglycaemia is inappropriate in the presence of hypoglycaemia!
Despite the new knowledge of pathogenesis, the management of the disease still presents the paediatrician with a choice of unsatisfactory treatments that have major long term implications for the child and his or her family.
Because of the urgent need to improve prognosis, the European network for research into hyperinsulinism (ENRHI) was created in 1997 through the support of the European Union. The network brings together leading basic and clinical scientists from seven European countries to encourage collaboration, generate new scientific data, and from these, to define a consensus guide to management.
A workshop was held in Finland in May 1999 to consider the latter, and this paper reflects the debate that occurred. As in any other area of medicine, consensus was achieved for many of the components of the management cascade. Nonetheless, controversy still surrounds other aspects, and these are defined and considered below. Further areas of clinical research that need to be developed are also highlighted.
Background
Hyperinsulinaemic hypoglycaemia has previously masqueraded under a variety of different descriptive names, including “idiopathic” hypoglycaemia of childhood, leucine sensitive hypoglycaemia, neonatal insulinoma, pancreatic microadenomatosis, nesidioblastosis, persistent hyperinsulinaemic hypoglycaemia of infancy, and congenital hyperinsulinism. Both sporadic and familial forms of the disease are recognised, the former having an estimated incidence in western Europe of one in 50 000 births.9 In isolated European communities, including parts of Finland, the disease incidence is much higher10; the highest incidence is found in societies with high rates of consanguinity.11 In these cultures, particularly in the Arabian peninsular, the incidence may be as high as one in 2500 births.10 ,11
Clinical presentation and immediate management
Most infants with hyperinsulinism present during the 1st postnatal days, with others during the 1st year. Rarely, older children present de novo with symptoms of hypoglycaemia. Figure 1 shows the age distribution in one large series of cases.12

Hyperinsulinism: age at presentation and surgery.
Hyperinsulinism can be associated with well defined clinical conditions.13-16 Thus, the Beckwith-Wiedemann syndrome manifests a transient hyperinsulinism and these babies have pathognomonic physical signs including exomphalos, macroglossia, and transverse creases of the ear lobes. This disease and the hyperinsulinism caused by maternal diabetes, rhesus incompatibility, perinatal asphyxia,13 and maternal glucose and drug treatment during delivery will not be considered further in this paper.
Infants with hyperinsulinism might have a characteristic appearance with macrosomia, strongly resembling the infant of a diabetic mother. The appearance suggests the occurrence of prenatal hyperinsulinism. However, not all infants have this appearance, and some of the most difficult management problems arise in infants born of normal or low birth weight, including preterm infants.17
The first clinical manifestations of hyperinsulinism include non-specific features such as “floppiness”, “jitteriness”, poor feeding, and lethargy. More dramatically, the infants may have seizures, coma, and even averted neonatal death.18
The importance of accurately measuring blood glucose in the presence of any of these symptoms cannot be overemphasised.
Considerable controversy surrounds the definition of neonatal and infantile hypoglycaemia.19-21 Pragmatically, any infant with a persistent measurement of blood glucose less than 2.6 mmol/litre (as measured by an accurate laboratory method and not a bedside screening test), particularly when accompanied by symptoms, should be the focus of particular attention, and the diagnosis of hyperinsulinism considered. Any persistent hypoglycaemia, whether symptomatic or not, needs investigation and treatment
Blood must be drawn and saved at the time of hypoglycaemia (together with the next urine sample passed) for assay of the substances listed in table 1.
Intermediary metabolites and hormones to be measured at the point of hypoglycaemia
A baby who cannot maintain normoglycaemia despite frequent enteral feeds (if necessary via a nasogastric feeding tube) needs to have the support of an intravenous glucose infusion to prevent neuroglycopenia. It should commence at the normal neonatal and infantile hepatic production rate of glucose; namely, between 4–6 mg/kg/min. The infusion rate should then be titrated upwards as necessary to maintain the blood glucose value above 2.6–3.0 mmol/litre.
The definition of glucose requirement to maintain normoglycaemia is a key diagnostic as well as therapeutic step. The demonstration of an increased glucose requirement is a powerful indicator of underlying hyperinsulinism.
Many children require infusion rates in excess of 15–20 mg/kg/min. Clearly, it is essential to document that the infant actually needs that high infusion rate. Infants are often referred to regional centres with alleged high glucose requirements, but on being challenged by a cautious decrease are found not to be so dependent. It is, of course, possible that the glucose dependency may change dramatically for the better in the 1st postnatal days, particularly in infants who may have experienced mild birth asphyxia.
The need to assess carefully and regularly the glucose infusion requirement without causing severe hypoglycaemia, symptomatic or not, is a key aspect of the initial management process.
In infants who are dependent on high rates of glucose infusion, the insertion of a central venous catheter through the umbilicus or via a peripheral vein is mandatory. This is because the sudden loss of a peripheral venous cannula in a child who has had multiple venepunctures can cause a rapid onset of dramatic symptomatic hypoglycaemia during attempts to re-site the line.
The need for early referral to a specialist centre
Participants at the consensus workshop unanimously criticised the current practice of late referral to a centre experienced in the management of hyperinsulinism, and urged earlier transfer.
Practitioners should not underestimate the practical difficulties of managing infants with hyperinsulinism, not least because of the volatility of glucose control and the vulnerability to unexpected and unpredictable severe hypoglycaemic episodes, but also because of the risks of fluid overload and sepsis contributing to the considerable morbidity encountered by these infants.
Of central importance is the need for dedicated nursing support delivered in a unit that has the infrastructure, quality standards, and training for rapid and repeated accurate blood glucose measurement. Protocols for the emergency management of the hypoglycaemic episodes need to be in place, and staff should have the experience and the knowledge to impart confidence to parents at a time of major emotional stress.
Members of ENRHI argue that late referral to a specialist centre after days or even weeks of fruitless attempts to make a diagnosis and establish effective treatment is one of the main reasons why the neurological morbidity remains so high. Indeed, because of the propensity of this disease to cause neural damage, it could be deemed negligent not to transfer early.
The transfer process itself needs to be carefully orchestrated, as outlined in table 2. Transfer without a secure intravenous line and a trained nursing or medical escort is not an acceptable practice.
Recommended guidelines to referring hospitals for transfer of babies with hyperinsulinism (transfer of babies/children with hypoglycaemia, regardless of aetiology)
Establishing the diagnosis and the cause of hyperinsulinism
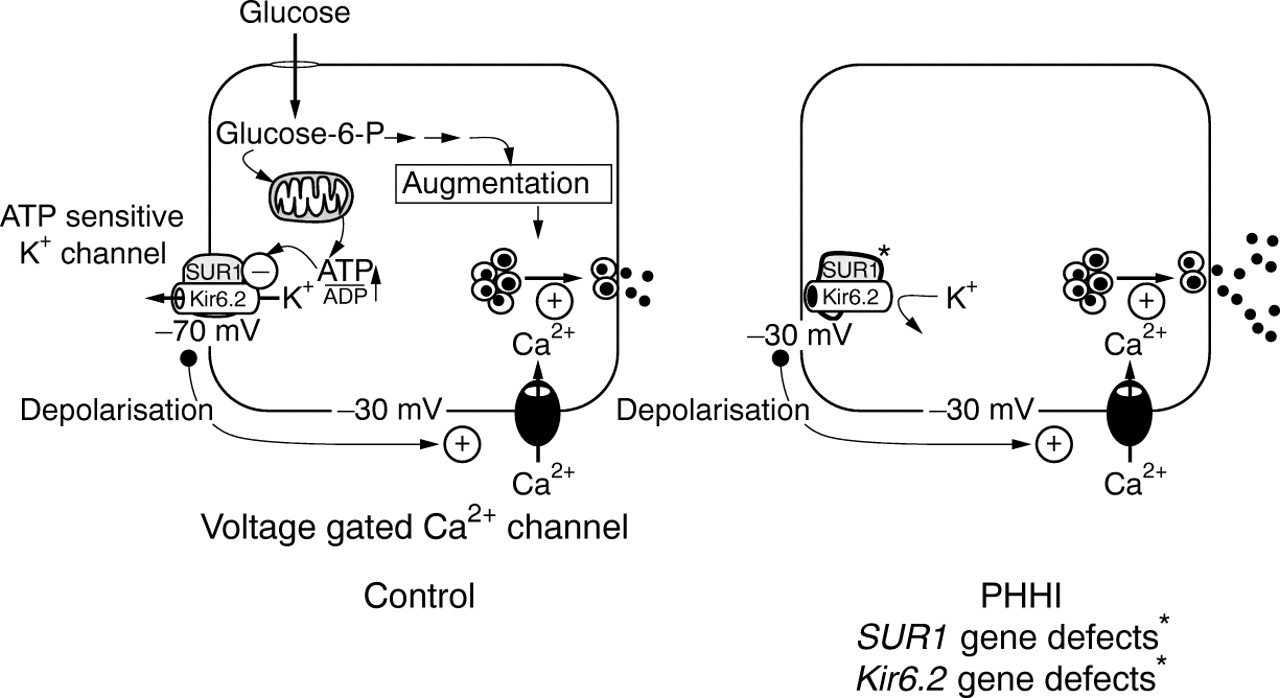
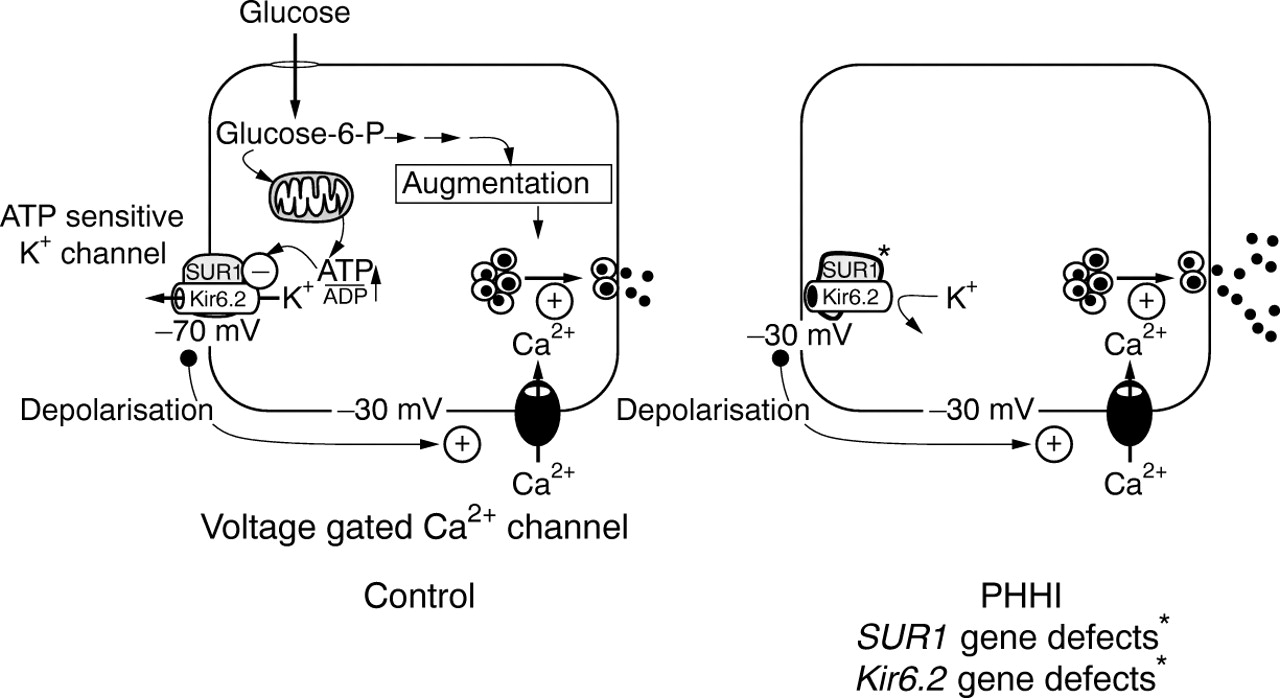
Figure 2 outlines the consensus model for the normal control of insulin secretion, and the loci at which abnormalities can occur. Knowledge of this simple schematic is essential to understand the rationale for the diagnostic and management cascades.
{kind=link}
{kind=link}
The sequence of events leading to the release of insulin from a pancreatic β cell. In brief, SUR1 and KIR6.2 constitute the ATP sensitive potassium (KATP) channel. These two proteins are encoded by genes located on chromosome 11p15.1. Mutations in these genes account for some cases of hyperinsulinism. The KATP channel is normally kept open maintaining a membrane resting potential of about –70 mV. When glucose enters the cell it raises the concentration ratio of ATP to ADP, which in turn causes the closure of the KATP channel. Once closed, the cell membrane becomes depolarised, allowing the influx of calcium through specific voltage gated calcium channels. This is the trigger for insulin exocytosis. Mutations have also been described in the genes for glutamate dehydrogenase (GDH) and in the glucokinase (GK) system. These defects increase the intracellular ATP to ADP ratio, thereby initiating insulin secretion. In these conditions, there is no abnormality in the potassium or calcium channel function. PHHI, persistent hyperinsulinaemic hypoglycaemia of infancy; glucose-6-P, glucose-6-phosphate. (Reproduced with permission from Shepherd et al22)
Three components of the diagnostic cascade need to be considered; namely, the biochemical diagnosis, the genotype and clinical phenotype, and the role of pancreatic imaging.
THE BIOCHEMICAL DIAGNOSIS
There is now a well established consensus on the primary and secondary diagnostic indicators for hyperinsulinism, as shown in table3. In many children, the diagnosis depends on the aggregation of several components, particularly because in the neonate the normal tight association seen in older children and adults between plasma insulin and blood glucose concentrations is not present.23 ,24 Attention is drawn to the following:
The characteristic metabolic profile of hypoketonaemic hypofattyacidaemic hypoglycaemia arises from the anabolic effects of insulin preventing metabolic counter regulation as the blood glucose concentration falls.
The presence of any measurable insulin during hypoglycaemia is strongly indicative of a defect in regulating basal insulin secretion in the face of a falling blood glucose concentration. The interpretation of the results depends on the sensitivity and specificity of the local insulin assay.
Hyperinsulinism can occur in the absence of florid hyperinsulinaemia—a normal insulin concentration for normoglycaemia is inappropriate in the presence of hypoglycaemia.
Insulin release is pulsatile and measurement of C peptide might be more helpful than a single measurement of insulin because it reflects overall insulin production; there is little extra value gained from measuring proinsulin concentrations.25
Very high glucose infusions rates (greater than 20 mg/kg/min) might be needed to secure normoglycaemia. Titration of the infusion rate against the blood glucose concentration achieved allows the impact of changes in treatment to be assessed.
Progressive hepatic enlargement, presumably as a result of glycogen deposition, might occur during high infusion rates of glucose, but also secondary to fluid retention as a result of high fluid infusion rates and diazoxide treatment.
Babies show a glycaemic response to glucagon injection when hypoglycaemic because of the mobilisation of glycogen deposits from the liver. This phenomenon can be of diagnostic as well as therapeutic benefit. There is a lack of consensus over the dose of glucagon used in different centres, ranging from 0.1 mg/kg to a single overall dose of 1.0 mg.26 In general, a positive response can be said to occur when there is a clear increment in blood glucose despite severe hypoglycaemia. The injection of glucagon can be useful at times of emergency, such as the re-siting of a venous cannula. Glucagon should be administered with caution because of its insulin stimulatory effect and the propensity to cause rebound hypoglycaemia.
Other markers of hyperinsulinism including low circulating levels of branched chain amino acids at the time of hypoglycaemia are of research interest only.27 The measurement of blood ammonia concentration is now mandatory in view of the recent description of the syndrome of hyperammonaemic hyperinsulinism.28 This condition is likely to be the explanation of the so called “leucine sensitive” hyperinsulinism in which children show sensitivity to protein ingestion.29
Plasma concentrations of cortisol and growth hormone might not be consistently raised during episodes of even severe symptomatic hypoglycaemia.30 Nonetheless, congenital hypopituitarism is an important differential diagnosis to consider; in this diagnosis, however, the plasma insulin concentration should be undetectable, and there may be other stigmata of the condition including mid line defect and, in boys, micropenis. If in doubt, specific pituitary stimulation tests should be performed at the tertiary referral centre.
An increase in the blood glucose concentration after somatostatin injection is suggestive of hyperinsulinism, but it is not recommended as a routine diagnostic criterion.
The diagnostic criteria for hyperinsulinism
ESTABLISHING THE GENOTYPE AND CLINICAL PHENOTYPE
Advances in the molecular genetics of the disease have shown hyperinsulinism to be caused, in about 30–50% of patients, by abnormalities in either genes controlling intracellular metabolic pathways or membrane cation transport. Most abnormalities described to date relate to the genes controlling the sulphonylurea receptor (SUR1) and the inward rectifier potassium channel (Kir6.2), proteins that together form the functional ATP dependent potassium channel (KATP) in the β cell membrane. Over 40 different mutations have been reported, most of which are recessively inherited.31 Dominantly expressed abnormalities have also been described.32 Although the molecular genetics of some of these cases are now known,33 there is no information on the pancreatic lesion in these patients because most patients with autosomal dominant hyperinsulinism do not require surgery.
Recently, a further twist has been added to the story of the genetics of hyperinsulinism, with the discovery that “focal” hyperplasia in the endocrine pancreas is the result of a unique combination of events: somatic loss of the maternal allele on the short arm of chromosome 11, in a patient harbouring an SUR1 mutation on the paternal allele.34 ,35 The juxtaposition of SUR1 and several imprinted genes on chromosome 11p15 appears to be responsible for this unique genetic mechanism of disease.
Gene mutations that cause abnormal activation of glucose signalling of insulin secretion by altering the enzyme kinetics of glutamate dehydrogenase or of glucokinase have been described recently.28 ,36 Other loci have yet to be defined in rare patients who present with hyperinsulinism in association with lactic acidosis.37 Because these abnormalities are not associated with abnormal KATP channels, most patients with these defects respond to diazoxide.
Despite these recent developments, less than 50% of patients studied to date have a definable genetic abnormality. Therefore, it is likely that other abnormalities will be revealed as the search continues for new defects in the secretory apparatus, including defects in the intracellular control of calcium signalling of exocytosis.38 For a more detailed description of the genetics of hyperinsulinism see Glaser et alelsewhere in this issue.39
Unfortunately it is not yet possible to use genetic analysis routinely to screen for mutations except in specific populations with unique and characteristic abnormalities usually associated with consanguinity.40
There is considerable variation in the clinical phenotype of severity in hyperinsulinism. In general, children with hyperinsulinism born to consanguineous parents, particularly those of Jewish or Arabic ancestry, have a severe form of the disease that is usually resistant to medical treatment. This also applies to patients from particular regions in Finland. It should be predicted that these patients are likely to require early surgery to control the hypoglycaemia. Siblings are also at risk of the disease and should be screened for the onset of neonatal hypoglycaemia.
In most European and white children, however, there is no family history of intermarriage or afflicted members. Moreover, with the exception of hyperammonaemic hyperinsulinism it is not possible to predict at presentation which infants will respond dramatically to diazoxide treatment, although in general, neonates presenting with severe hypoglycaemia in the 1st postnatal days have the most unresponsive form. It is equally impossible to predict which infants will have “transient” as opposed to “persistent” hyperinsulinism. In general, children with hyperammonaemic hyperinsulinism and upregulation of glucokinase have a milder disease phenotype, which usually responds rapidly to moderate doses of diazoxide, but a genetic heterozygosity certainly exists.
IMAGING OF THE PANCREAS
The need for methods to identify “focal” areas of hyperplasia has been given new impetus through the reports that preoperative percutaneous trans-hepatic pancreatic venous sampling can identify “hot spots” of insulin hypersecretion.41 In children, preoperative pancreatic venous sampling should be performed under general anaesthesia in an interventional angiographic room. Care should be taken to maintain blood glucose concentrations at between 2.5 and 3 mmol/litre. The right portal vein is punctured with a 21 gauge catheter needle. Specially shaped 3F catheters are then exchanged over a guide wire. Multiple venous samples are taken in the splenic, mesenteric, and portal veins but also in the small pancreatic veins draining the head, the body, and the tail of the pancreas. The concentrations of insulin, glucose, and C peptide are mapped according to the venous anatomy of the pancreas. A focal form is diagnosed when a single or two contiguous veins have high amounts of insulin. The C peptide to insulin ratio is close to one in those veins in that setting. A diffuse form is diagnosed when several non-contiguous veins have high concentrations of insulin. In cases of high concentrations of glucose the mapping is non-contributive. In a few patients, despite a good control of glucose below 3 mmol/litre, all the sampled veins show low or normal concentrations of insulin. These patients correspond to focal forms in which the draining vein of the lesion has not been selectively sampled. When followed by meticulous intraoperative microdissection, aided by immediate histological confirmation of hyperplasia, there is evidence that the long term prognosis is much better than in patients subjected to blind 95% partial pancreatectomy.41 Furthermore, focal disease might occur only in the head of the pancreas, and this may not be resected with blind partial pancreatectomy.
The ability to identify focal endocrine hyperplasia is a new and major advance in diagnosis and management. However, the logistic aspects of this approach are substantial in terms of the availability of staff experienced in interventional radiology and in pancreatic histology. Moreover, expert nursing care is needed because the infants are at serious risk of hypoglycaemia because medical treatments need to be discontinued for several days before the procedure. In addition, the imaging procedure requires that the infant's blood glucose be brought as close as possible to hypoglycaemic values for up to several hours. The technique requires further evaluation before it can be recommended for general use. Moreover, because of the logistic problems, the consensus workshop recommended that the use of the technique should be restricted to one or two national centres in each country that have the comprehensive resources needed to manage the children competently.
Alternative means have been proposed for identifying focal disease. These include the intra-arterial calcium stimulation test, which is based on the insulin stimulatory effect of rapid selective injection of calcium into the branches of the coeliac arteries supplying the different parts of the pancreas.42 This technique also requires further evaluation, with members of the consensus workshop expressing concern over the potential for infarction of the coeliac arteries.
Other techniques, including preoperative and intraoperative direct pancreatic ultrasonography, computed tomography, magnetic resonance imaging, and coeliac angiography have been used by many investigators. The limited published information has not shown these techniques to be of value, particularly in the neonate.43 However, imaging should be considered in the older child in whom the possibility of the presence of an isolated adenoma is high.1
The consensus workshop participants agreed that currently available imaging techniques were not of value in the routine investigation of neonatal and early infantile hyperinsulinism; they should only be considered in the search for an insulinoma in an older child presenting with recent onset of hypoglycaemia.
Management
It is important at the outset to identify the objectives of management. They are to: (1) prevent hypoglycaemic brain damage and allow normal psychomotor development; (2) establish normal feed volume, content, and frequency for the age of the child; (3) ensure normal tolerance to fasting for age without developing hypoglycaemia; and (4) maintain family integrity.
A regimen cannot be said to be successful if it does not fulfil these objectives. However, in practice it might be very difficult to achieve them.
MEDICAL TREATMENT
Initial stabilisation
The mainstay of initial medical treatment is the provision of adequate carbohydrate to maintain blood glucose concentrations above 2.6–3.0 mmol/litre. Very high infusion rates of glucose (> 20 mg/kg/min) in addition to frequent enteral feeds may be required. This might demand the insertion of a central venous catheter to allow the administration of glucose in high concentrations, together with a nasogastric feeding tube for regular feeds. Glucose polymer can be added to the enteral feed to increase the carbohydrate intake. However, it is important not to cause too great an osmolar load in the gastrointestinal tract that might predispose to the onset of necrotising enterocolitis. Because of the immaturity of intestinal amylase during the 1st year of life, the administration of cornstarch might not be effective.
When the blood glucose concentration has been stabilised, pharmacological agents need to be introduced to try to decrease insulin secretion and normalise the carbohydrate intake. The impact of their introduction can be tested by the definition of a change in glucose requirement. It is recommended that these manoeuvres be attempted only in referral centres experienced in their use.
Pharmacological treatments
A logical progression through the available treatments is proposed (table 4), which allows a clear definition of the clinical phenotype. The essential principle of management is to introduce only one change in dose or treatment at a time and to assess the impact before moving on to the next change.
Drugs used in the medical management of hyperinsulinism
The drugs of first choice are those that can be given orally, because some children may respond very well, even if they require ongoing treatment for several months or years. It is reasonable to argue that it is essential to prove that a child will not respond to such agents before introducing more powerful hormones.
Diazoxide (10–20 mg/kg/day in two to three divided doses) and chlorothiazide (7–10 mg/kg/day in two divided doses) are recommended for initial treatment and should always be given together. Both these agents activate potassium channels by different mechanisms,6 the diuretic also being given concurrently for its ability to overcome the fluid retaining effects of diazoxide. There is controversy over the frequency with which infants respond fully to these drugs, with reports ranging from 15% to 60%, or more.12 ,41 ,44 This difference in responsiveness could perhaps reflect the selection of cases being referred to the centres. There is little information on the pharmacodynamics or pharmacokinetics of diazoxide in childhood. Children who fail to respond to a dose of 15–20 mg/kg/day might have pancreata that will respond in vitro to higher concentrations. In practice, the limit of clinical tolerance is determined by the magnitude of side effects, in particular fluid retention and cardiac failure, and most children who will respond do so at doses up to 15 mg/kg/day.
It is logical, in addition to activating potassium channels by diazoxide and chlorothiazide, to try to decrease voltage dependent calcium channel activity by blocking agents, such as the slow release form of nifedipine at a dose of 0.25–2.5 mg/kg/day.17The clinical response to this drug is highly variable, an observation that has now been explained by studies performed in vitro on β cells from patients with persistent hyperinsulinaemic hypoglycaemia of infancy.38 Thus, some children with KATPchannel abnormalities might have a concurrent defect in voltage gated calcium channels. It is not possible to predict which children will respond without trying the drug. The drug has not proved as useful as was first hoped, yet nifedipine may still have a role as an adjuvant, although the long term effects of continuous treatment are unknown. The drug is light sensitive and there are practical difficulties in its formulation when used in small children. Further research is needed to define the precise value and role of calcium channel blocking drugs; nonetheless, their use is one tangible benefit that has arisen from a better understanding of the pathophysiology of the β cells in this condition.
The side effects of these drugs are important. Diazoxide causes fluid retention that can precipitate cardiac failure. Rarely, it causes a blood dyscrasia. The most distressing effect from the parental viewpoint is the stimulation of generalised hypertrichosis. Nifedipine appears to be remarkably safe in the doses used, but blood pressure monitoring is mandatory.
Second line agents are those that need to be given by infusion or injection. Ideally, they should be given when the orally administered drugs have been shown not to be effective, particularly if the child remains glucose infusion dependent. In practice, however, there may be compelling reasons to begin them because of the enormity of the management problems in controlling the hypoglycaemia.
The two most important substances are glucagon and somatostatin. There is a consensus that both hormones are of value, but controversy over the doses used and the need to give them singly or in combination. Both hormones when given in high doses cause tachyphylaxis.
Glucagon has a powerful effect on mobilising glucose from hepatic glycogen.1 Its administration by means of a continuous intravenous infusion at rates of between 5 and 10 μg/kg/hour can help reduce the infusion rate of glucose needed to maintain normoglycaemia. Some authorities recommend its early use to gain rapid stabilisation of blood glucose concentrations; others argue that it is a powerful insulin secretagogue, and its administration in theory will maintain a drive to insulin hypersecretion. For this reason, they propose using glucagon only with the concurrent administration of somatostatin.
The somatostatin analogue, octreotide, activates potassium channels in the β cell membrane, and may also affect the intracellular translocation of calcium.7 The doses proposed for initiating combination treatment are 1.0 μg/kg/hour of glucagon together with 10 μg/kg/day of octreotide.6 ,7 It should be noted that these doses are less than those advocated for single hormone infusion. Further systematic research is needed to define the effects and role of these powerful hormones.
The use of steroids remains a controversial issue. In general, high doses of steroids have been contraindicated. However, the recent demonstration of cortisol unresponsiveness during hyperinsulinaemic hypoglycaemia30 raises important questions for further research, not only on the mechanism of this observation, but also on the impact of introducing cortisol substitution. Somatostatin administration leads to the inhibition of a number of other hormone systems,45 and in some centres such children are given hydrocortisone replacement, particularly during surgical procedures. This approach also needs further investigation and validation.
FURTHER MANAGEMENT IN MEDICALLY RESPONSIVE CHILDREN
The overall aim of medical treatment is to prevent hypoglycaemia while allowing a normal feeding pattern to be established. Children who respond rapidly to diazoxide treatment (as demonstrated by an ability to withdraw glucose support) should be maintained on their initial dose with repeated systematic attempts to withdraw the diazoxide during the next few months. Some children, particularly those with hyperammonaemic hyperinsulinism, might require treatment for many years. Others, without hyperammonaemia, might also remain diazoxide dependent until adulthood. Attempts to withdraw diazoxide should only be made in hospital.
In severe early onset hyperinsulinism, there is a tendency for gradual improvement in the clinical severity of the defect with the passage of time. This observation led Glaser et al to adopt an aggressive medical treatment strategy comprising long term subcutaneous glucagon and octreotide infusions and continuous overnight gastrostomy feeds.46 The approach has been used with some success in children with the Ashkenazi Jewish and Palestinian Arab phenotype of hyperinsulinism.47 The part that this approach should play in treating other populations is still to be defined, and the short and long term effects of this powerful hormonal treatment need further evaluation. Many families find the intensity of the regimen impossible to cope with in their daily lives.
FAILURE OF MEDICAL TREATMENT: SURGICAL TREATMENT
Children who fail to respond to medical treatment should be managed in a centre where there is the closest of collaboration and dialogue between the medical and surgical teams. Some experts recommend that aggressive medical treatment should be continued for at least four to six weeks in early onset hyperinsulinism. Hypoglycaemia must be prevented during this period, through a combination of high carbohydrate administration rates and drugs, as described above. The rationale is that an improvement in the severity of the hyperinsulinism may be seen during this period; however, there are no published studies that evaluate the change in the need for surgery in infants in whom medical treatment is initially unsuccessful at two to three weeks compared with five to six weeks. The criteria for successful medical management are a feeding regimen acceptable to the family, with normal blood glucose concentrations during reasonable periods of fasting. If an acceptable regimen for home oral feeding cannot be established without hypoglycaemia, a surgical approach to management should be considered.
Because of the risk of major management related complications, including central line sepsis, venous thrombosis, hepatic dysfunction, bleeding diatheses, impaired nutrition, and hypoglycaemic encephalopathy, the consensus view expressed by the workshop was that children should be subjected to surgery sooner rather than later.
The absolute indications for surgery are: (1) demonstration of focal hyperplasia in children unresponsive to medical treatment; and (2) glucose infusion dependency despite maximum doses of diazoxide, chlorothiazide, nifedipine, glucagon, and somatostatin.
Infants with diffuse disease will normally require a 95% pancreatectomy to control the hyperinsulinism. After this procedure there is a high incidence of pancreatic endocrine and exocrine insufficiency. In infants with focal forms of the disease, an opportunity exists for a partial pancreatectomy, preserving the normal pancreatic tissue. The focal areas of abnormal pancreas are usually not macroscopically apparent at surgery and their resection requires skilled microdissection aided by immediate histological examination of the resected tissue.41 Confirmation of the histological diagnosis is by frozen section, the distinction between focal and diffuse forms being made on the basis of nuclear size and nuclear crowding.48
The routine performance of 95% pancreatectomy in all cases of persistent hyperinsulinaemic hypoglycaemia of infancy coming to surgery is no longer justifiable. The new standards in treatment will require the centralisation of management to a small number of designated centres with the necessary resources and expertise.
POSTOPERATIVE MANAGEMENT
Early postoperative glucose intolerance
A reduction in glucose requirement is normally evident during surgery. Infants will frequently become hyperglycaemic during the immediate postoperative period. To avoid massive swings in blood glucose concentrations, these infants should be changed to 5% dextrose infusions, with insulin being given with a sliding scale regimen. These infants are usually exquisitely sensitive to exogenous insulin. The insulin requirement will normally diminish early in the postoperative course, and the optimum outcome is to be able to discontinue all treatment. The ability to do so is greatest in children with focal disease.41
Postoperative diabetes
The risk of developing diabetes mellitus later on in childhood after a 95% pancreactectomy is high.49 ,50 Interestingly, these children exhibit some unusual characteristics in comparison with children who have type 1 juvenile diabetes mellitus.51 For example, they have striking resistance to hyperketonaemia and diabetic ketoacidosis. Management of the secondary diabetes involves using either an intermediate acting insulin or using a combination of short and long acting insulins in conjunction with dietary advice. Furthur studies are needed to find out whether these children have a similar chance of developing the serious complications of diabetes to children with “ordinary” diabetes and whether the magnitude of these complications is the same or different.
Postoperative pancreatic exocrine insufficiency
Studies of exocrine pancreatic function in infants who have undergone 95% pancreatectomy suggest that exocrine function is compromised from the early postoperative period, although malnutrition sufficient to cause growth failure may not be apparent for a variable period (Lindley K, unpublished, 1999). Evidence of impaired pancreatic exocrine function is sought by measurement of stool elastase. Infants with decreased concentrations should have pancreatic enzyme replacement.
Feeding management
Foregut dysmotility and gastro-oesophageal reflux is common in hyperinsulinism.52 The pathophysiology is yet to be determined. Infants may show poor sucking and swallowing, retching, vomiting, and intestinal dilatation; feeding problems are compounded by the use of nasogastric or gastrostomy feeds, which delays the establishment of normal feeding patterns, taste, and “orality”. In severe cases, the deprivation of oral stimulation may require months of rehabilitation with skilled speech and language therapists. Infants in whom severe gastro-oesophageal reflux is demonstrated preoperatively should undergo an antireflux surgical procedure at the same time as having pancreatic surgery (Nihoul-Fekete C, personal communication, 1999). This approach and the use of gut motility agonists including erythromycin requires formal evaluation. The intestinal dysmotility problems are especially difficult to manage in children from the Arabian peninsular, many of whom require months of hospital management.
Neurological management
It is unfortunate that so many infants with hyperinsulinism develop evidence of neurological damage, ranging from the subtle to the severe.2 ,3 ,53 ,54 Careful repeated documentation of neurological status is mandatory; this might require expert paediatric neurological investigation, including magnetic resonance imaging and management of complex disabilities.
Other aspects
In children subjected to pancreatic surgery, postoperative imaging of the spleen is needed to document splenic integrity and the absence of the need for long term penicillin prophylaxis against pneumococcal infections.
Further hypoglycaemia
A small number of infants remain severely hypoglycaemic despite 95% pancreatectomy. Possible reasons include the presence of focal lesions in the head of the pancreas or diffuse disease that is medically unresponsive. The management of this circumstance demands further sequential introduction of medical treatment, and consideration of total pancreatic resection. After an initial period of good control, some infants develop hypoglycaemia after weeks or months. This is usually a result of regeneration of the pancreatic remnant for reasons that are not fully understood.
Psychosocial dimensions
The management of hyperinsulinism imposes intolerable burdens on the families of the children as they witness weeks of difficult treatment and the infliction of pain on their infants. It is hardly surprising that many fail to cope and disintegrate under the strain. Management demands skilled psychological and social support.
As the children become older, they may rebel against the need for insulin treatment and become highly manipulative in the face of substantial if not dominating family preoccupations with the risk of hypoglycaemia.
Natural history
Studies of predominantly Ashkenazi Jewish children with hyperinsulinism suggest that the natural history of the disease is one of progressive glucose intolerance and clinical diabetes. By undertaking both glucose tolerance tests and hypoglycaemic clamp studies it has been shown that individuals with frankly diabetic glucose tolerance tests have evidence of persistent inappropriate insulin secretion in the face of hypoglycaemia.44 This suggests that such individuals are diabetic by virtue of having a reduced mass of functionally abnormal β cells. The diabetic phenotype might be the result of enhanced apoptosis in hyperinsulinaemic β cells.
The future
The important distinction between focal and diffuse disease has been highlighted throughout this commentary. Current methods of diagnostic evaluation are cumbersome and difficult. The search for other techniques that are less invasive must be of high priority. The role of genetic analysis and improvements in identifying the genome in the diagnostic investigation are also areas for further reasearch, not least because so many children at present have not been shown to possess a known mutation.
New pharmacological agents with which to inhibit insulin secretion are urgently needed. Long acting octreotide and insulin specific analogues are available from pharmaceutical companies, but are either not licensed for human use, or only for adults.
Finally, the proof of principle that β cells from some resected pancreata continue to replicate in tissue culture, coupled with the ability to affect gene therapy in vitro, offers a tantalising future prospect of reimplanting the patient's own genetically modified cells that have regained normal glucose dependency of insulin secretion.55
Acknowledgments
The European network for research into hyperinsulinism is supported by a concerted action grant (Biomed 2 programme: grant number BMH4-CT98–3284) from the European Commission. Some of this work was undertaken by Great Ormond Street Hospital for Children NHS Trust who received a proportion of its funding from the NHS Executive; the views expressed in this publication are those of the authors and are not necessarily those of the NHS Executive.