Article Text

Abstract
We report on a child in whom severe nutritional vitamin B12 deficiency was exacerbated by a genetic impairment of the folate cycle, causing reduced CSF concentrations of the methyl group donor 5-methyltetrahydrofolate. Some patients with vitamin B12 deficiency may benefit from high dose folic acid supplementation, even if plasma concentrations are high.
- vitamin B12 deficiency
- MTHFR polymorphism
- folate cycle
- CSF, cerebrospinal fluid
- MTHF, 5-methyltetrahydrofolate
Statistics from Altmetric.com
The interaction between environmental factors and genetic variants is known to determine disease manifestation in individual patients. We present a case which illustrates the value, and potential therapeutic relevance, of genetic analyses in an apparently straightforward exogenous disorder such as vitamin deficiency.
CASE REPORT
A 15 month old boy presented with coma, muscular hypotonia, and dehydration. He was the first child of a mother on a strict vegetarian diet and was still fully breast fed; the mother was again 11 weeks pregnant. Development had reportedly been normal until the age of 13 months when the boy became increasingly sleepy, lethargic, and a poor feeder. On admission he was malnourished (weight 7600 g, 900 g below the third percentile) with height and head circumference in the normal range. There was severe metabolic acidosis and macrocytic anaemia (haemoglobin after rehydration 48 g/l, MCV 101 fl). EEG showed continuous subdelta or delta activity; cranial nuclear magnetic resonance imaging revealed subcortical and cortical brain atrophy. Plasma vitamin B12 was low (85 pg/ml, ∼63 pmol/l; norm 232–1132 pg/ml), folic acid high (19.2 ng/ml, ∼44 nmol/l; norm 2.9–16.9 ng/ml). Metabolic investigations showed increased total homocysteine in plasma (65 μmol/l, norm <8), reduced methionine in plasma (9 μmol/l, norm 15–35), and cerebrospinal fluid (CSF) (3 μmol/l, norm 4.5–6.5), and methylmalonic aciduria (132 mmol/mol creatinine, norm <10). The CSF concentration of 5-methyltetrahydrofolate (MTHF) was reduced (25 nmol/l, norm 30–180). Analysis of the 5,10-methylenetetrahydrofolate reductase (MTHFR) gene revealed homozygosity for the common mutation A222V (frequently denoted 677C→T). The plasma vitamin B12 concentration was decreased in the mother (183 pg/ml, ∼135 pmol/l).
Treatment with hydroxycobalamin 1 mg intramuscularly (twice, followed by an oral multivitamin preparation) led to highly increased plasma vitamin B12 concentrations and normalisation of blood picture, plasma homocysteine, and urinary methylmalonic acid within one week. However, neurological recovery was slow. The boy started to fixate after six days of treatment, moved actively after 12 days, gained head control after three weeks, started to sit and vocalise after seven weeks, and could stand with support after 12 weeks. EEG returned to normal after five weeks; nuclear magnetic resonance imaging after seven weeks showed improvement of brain atrophy. Developmental status at age 22 months resembled that of a 12–15 month old child.
DISCUSSION
The severe and prolonged neurological disease in our patient may be explained by the combined effect of vitamin B12 (cobalamin) depletion and folate cycle disruption. Nutritional vitamin B12 deficiency has long been recognised as a cause of severe encephalopathy in breast fed infants of mothers on a vegetarian diet.1,2 The vitamin is exclusively obtained from animal sources, and hepatic stores in infancy may fall rapidly with insufficient supply. Vitamin B12 may be low in breast milk of vegetarian mothers, and this may have been exacerbated in our patient (who was comparatively old at the time of presentation) by the new pregnancy of the mother; maternal vitamin B12 concentrations are known to decrease during pregnancy.3
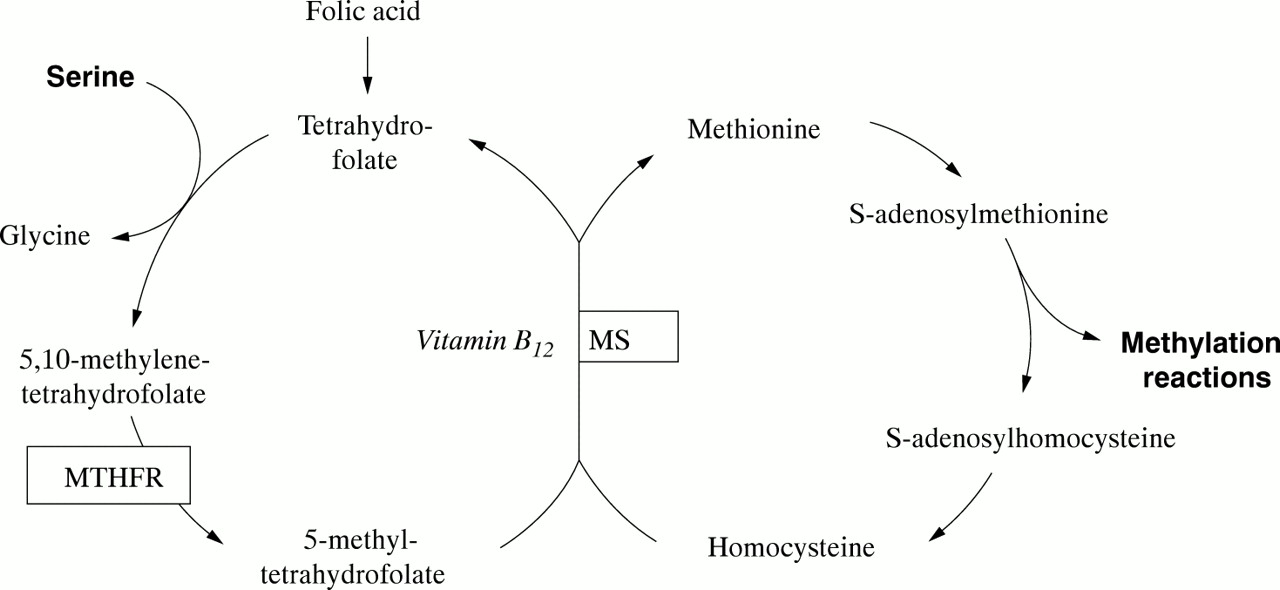
Children with nutritional vitamin B12 deficiency develop normally in the first months of life but subsequently show progressive lethargy, muscular hypotonia, loss of acquired skills, and coma. Neurological damage is thought to be a result of insufficient availability of S-adenosylmethionine, the principal methyl group donor in cellular metabolism (fig 1). Typical laboratory findings include macrocytic anaemia, hyperhomocysteinaemia caused by reduced activity of vitamin B12 dependent methionine synthase, and methylmalonic aciduria caused by reduced activity of vitamin B12 dependent methylmalonyl-CoA mutase. Most children improve rapidly after vitamin B12 supplementation.1
{kind=link}
Vitamin B12 (methylcobalamin) is a cofactor in the methylation of homocysteine to methionine, a central step in the cytosolic transfer of 1-carbon units from serine to S-adenosylmethionine. MTHFR, 5,10-methylenetetrahydrofolate reductase; MS, methionine synthase.
The slow neurological recovery in our patient is unusual and appears to be caused by concomitant mild MTHFR deficiency that exacerbated the disruption of the folate cycle. It is well established that vitamin B12 deficiency causes folate to become trapped as MTHF, as has also been reported in methionine synthetase deficiency.4 We expected, therefore, to find an increased CSF concentration of MTHF in our patient. Instead, MTHF was notably reduced, indicating an independent second pathogenetic factor.
The MTHFR polymorphism A222V5 (allele frequency 0.32 in whites6) causes reduced enzyme activity and MTHF availability and is associated with hyperhomocysteinaemia, vascular disease, and neural tube defects in homozygous individuals. Approximately 10% of the general population are homozygous for A222V and thus at risk for adverse effects under certain conditions. It is important to note that plasma folic acid in our patient (as well as previously described children with nutritional vitamin B12 deficiency2) was increased. Cerebral folate cycle disruption was evident only in reduced MTHF concentration in CSF, a result which became available only weeks after the start of treatment. In retrospect we believe that high dose folic acid supplementation (in addition to cobalamin) may have benefited our patient by increasing the intracellular availability of MTHF as methyl group donor, although this will need to be confirmed in future patients.
In conclusion, detailed characterisation of cerebral folate status should be considered in patients with nutritional vitamin B12 deficiency and severe neurological symptoms, even if plasma concentrations of folic acid are high. Mutation analysis for the common MTHFR polymorphism A222V, which is rapid, relatively inexpensive, and now widely available, may be helpful in cases in which CSF investigations are not possible. Further study of paediatric and adult patients with nutritive vitamin B12 deficiency is required to confirm a correlation between severity of neurological symptoms and genetic variants of the folate cycle.
Acknowledgments
We wish to thank Dr Henk Blom for helpful comments on an earlier draft of this paper.