Article Text

Abstract
Objective Neonatal-onset mitochondrial disease has not been fully characterised owing to its heterogeneity. We analysed neonatal-onset mitochondrial disease in Japan to clarify its clinical features, molecular diagnosis and prognosis.
Design Retrospective observational study from January 2004 to March 2020.
Setting Population based.
Patients Patients (281) with neonatal-onset mitochondrial disease diagnosed by biochemical and genetic approaches.
Interventions None.
Main outcome measures Disease types, initial symptoms, biochemical findings, molecular diagnosis and prognosis.
Results Of the 281 patients, multisystem mitochondrial disease was found in 194, Leigh syndrome in 26, cardiomyopathy in 38 and hepatopathy in 23 patients. Of the 321 initial symptoms, 236 occurred within 2 days of birth. Using biochemical approaches, 182 patients were diagnosed by mitochondrial respiratory chain enzyme activity rate and 89 by oxygen consumption rate. The remaining 10 patients were diagnosed using a genetic approach. Genetic analysis revealed 69 patients had nuclear DNA variants in 36 genes, 11 of 15 patients had mitochondrial DNA variants in five genes and four patients had single large deletion. The Cox proportional hazards regression analysis showed the effects of Leigh syndrome (HR=0.15, 95% CI 0.04 to 0.63, p=0.010) and molecular diagnosis (HR=1.87, 95% CI 1.18 to 2.96, p=0.008) on survival.
Conclusions Neonatal-onset mitochondrial disease has a heterogenous aetiology. The number of diagnoses can be increased, and clarity regarding prognosis can be achieved by comprehensive biochemical and molecular analyses using appropriate tissue samples.
- neonatology
- data collection
- mortality
- statistics
Data availability statement
Data are available on reasonable request. Deidentified participant data are available from corresponding author, Kei Murayama. kmuraya@mri.biglobe.ne.jp. ORCID iD 0000-0002-3923-8636. Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
What is already known on this topic?
Neonatal-onset mitochondrial disease has not been fully characterised due to its heterogeneity.
The number of molecularly diagnosed neonatal-onset mitochondrial disease cases is increasing, many of which are derived from nuclear genes.
Non-specific manifestations such as intrauterine growth retardation, early postnatal decompensation and poor feeding suggest the possibility of neonatal-onset mitochondrial disease.
What this study adds?
This observational study of neonatal-onset mitochondrial disease in the largest cohort ever is the first in Asia and shows more diverse genetic aetiology than that previously reported.
Combining a mitochondrial respiratory chain enzyme assay with cellular oxygen consumption rate measurement increases the number of molecular diagnoses of neonatal-onset mitochondrial diseases.
In neonatal-onset mitochondrial diseases, diagnosis of Leigh syndrome and confirmed molecular diagnosis have independent and significant effects on the survival curve.
Introduction
Mitochondrial diseases (MDs) occur in all age groups and present with several clinical features. However, cases of neonatal-onset follow different courses from those that begin in infancy or adulthood, with many of them becoming serious. Therefore, the time of onset should be taken into account in clinical classification to distinguish neonatal-onset from postneonate-onset MD. To date, the clinical features of MD with onset within 28 days of birth have been reported.1 2
However, studies that include a large number of patients with neonatal-onset MD are limited, and its clinical and molecular features have not been fully elucidated due to the heterogeneity of MD. In addition, with the increasing number of molecularly diagnosed cases,3 the case background is predicted to be different from that previously reported. Here, in a large retrospective observational study in Japan, we analysed 281 patients with neonatal-onset MD diagnosed by both biochemical and genetic approaches to clarify its clinical features, molecular diagnosis and prognosis. The clinical course and the results of biochemical and molecular analyses were used to classify disease type and compare them statistically to find a trend in heterogeneity. To the best of our knowledge, there are no reports on factors affecting the prognosis of neonatal-onset MD; this is the first statistical analysis of these factors.
Methods
We performed a retrospective analysis of patients diagnosed with neonatal-onset MD. According to previous reports,1 2 neonatal-onset MD was defined as the onset of disease within the first 28 days of life. Patients were suspected to have MD based on clinical findings according to the clinical section of the major and minor diagnostic criteria described by Bernier et al 4 and were diagnosed using biochemical and genetic approaches. For the biochemical approach, we first checked for decreased mitochondrial respiratory chain (MRC) enzyme activity. If there were no changes, we measured the cellular oxygen consumption rate (OCR) to check for mitochondrial dysfunction. The genetic approach was used when a sibling was confirmed to have MD based on molecular diagnosis or when there were no samples available for biochemical diagnosis. Written informed consent was obtained from the parents of all patients included in the study.
Patients
In total, 1023 patients were diagnosed with MD from January 2004 to March 2020, 294 (28.7%) of whom had neonatal onset. Excluding 13 cases whose clinical course was unknown, 281 patients were enrolled in this study.
Disease types
Patients were divided into four categories: multisystem MD (MsMD), Leigh syndrome (LS) cardiomyopathy (CM) and hepatic disease (HD). MsMD included patients primarily associated with multiple organ failure and hyperlactataemia that could not be classified as LS, CM or HD. LS was diagnosed using the criteria defined by Thorburn et al.5 Patients recognised to have Leigh-like syndrome by Thorburn’s definition5 were also included in the LS group. CM/HD included patients with CM/hepatic disorder as the primary symptom, with or without other mild systemic symptoms specific to MD and were diagnosed with mitochondrial dysfunction using the myocardial6/liver7 tissues and/or a molecular analysis.8 9
MRC enzyme assay
MRC enzyme activity was determined using skeletal muscle, the liver, the myocardium and skin fibroblasts, as previously described.10 MRC impairment was confirmed according to the minor diagnostic criteria described by Bernier et al,4 namely an activity of 30% or less of any RC complex in a tissue or less than 40% of the activity of any RC complex in a cell line.
OCR measurement
OCR was examined using skin fibroblasts as previously described.11 Mitochondrial dysfunction was defined as a maximum respiration rate of ≤71.6% of the normal rate.12
Variant analysis of the genes responsible for MD
Genomic DNA from biopsy or autopsy samples was extracted and analysed by Sanger sequencing, whole mitochondrial DNA sequencing3 and targeted resequencing of whole mitochondrial DNA. The exons of nuclear-encoded genes that cause MDs13 were analysed by whole-exome sequencing,3 14 or high-density oligonucleotide arrays to identify large chromosomal deletions.3
Statistical analysis
Categorical variables are presented as count and percentage. Multiple comparisons with the Fisher’s exact test using the Bonferroni method were performed to compare categorical variables among three to four groups. A multivariate analysis was performed to identify independent predictors for all-cause mortality. The starting point for survival analyses was the date of birth, and the censoring point was the date of death or the end of the follow-up period. Survival curves were generated following Kaplan-Meier analysis. Log-rank tests based on the occurrence of preterm birth, small-for-gestational-age (SGA), hyperlactataemia, early onset (within 2 days of birth), molecular diagnosis, MD with specific treatment options15 and each disease type were performed to determine the differences in survival rates. These factors were selected from clinical suspicion cases and previous reports.16 Cox proportional hazards regression analyses were performed on factors significant at p<0.10 in the log-rank test. All statistical tests were two sided, and results with p<0.05 were considered statistically significant. Statistical analyses were performed using EZR (Saitama Medical Center, Jichi Medical University, Saitama, Japan).17
Results
Breakdown of subjects by a diagnostic approach
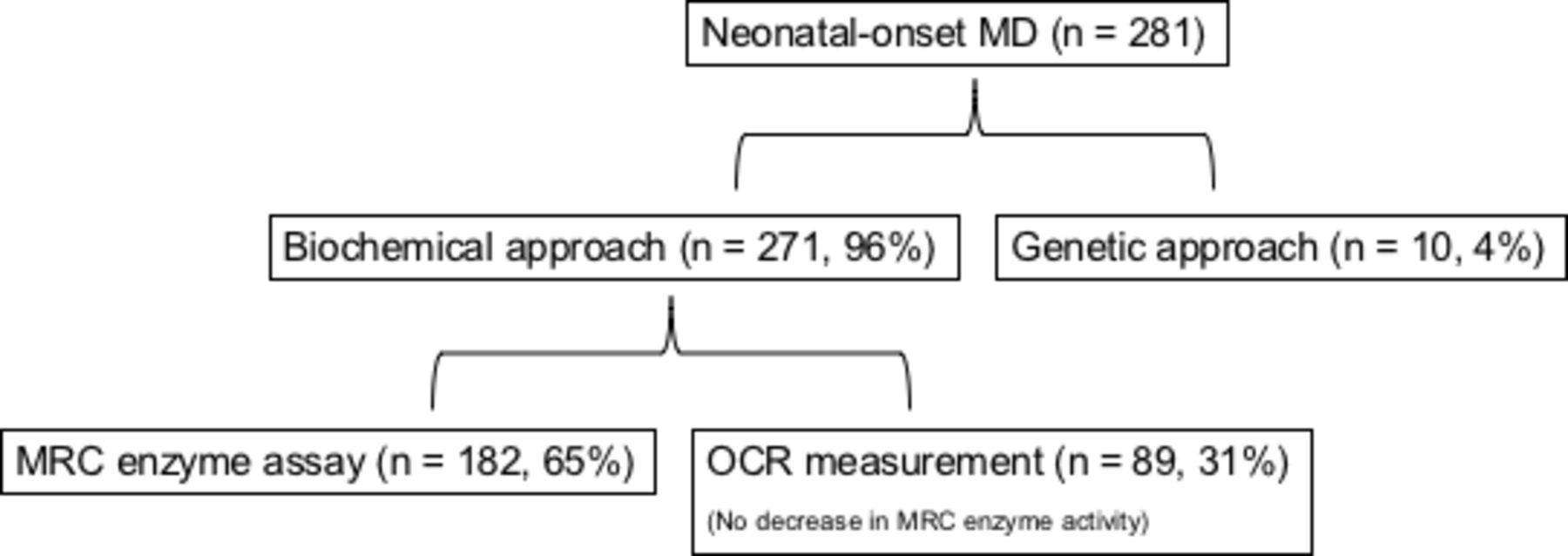
Figure 1 shows the breakdown of patients. MD diagnosis was made in 271 (96.4%) patients using the biochemical approach and in 10 (3.6%) patients using the genetic approach. Of the patients diagnosed using the biochemical approach, 182 (64.8%) had decreased MRC enzyme activity; 89 (31.7%) patients who did not have decreased MRC enzyme activity had decreased OCR.
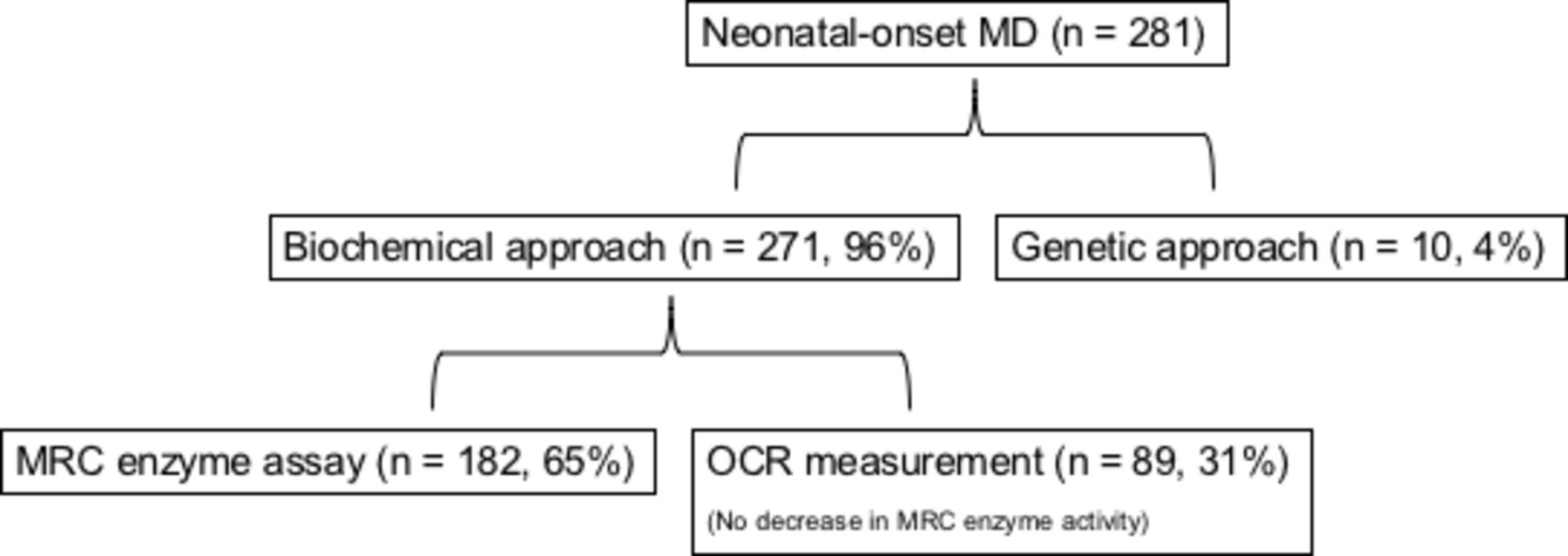
Breakdown of subjects using the diagnostic approach. Cases diagnosed by oxygen consumption rate (OCR) using the biochemical approach are those without decreased mitochondrial respiratory chain (MRC) enzyme activity in obtained samples. MD, mitochondrial disease.
Clinical features
Table 1 shows a summary of the patient cohort, comprising 281 patients from 276 non-consanguineous families. Of patients with available information, the mean birth weight was 2346±801 g (range: 422–4168 g, n=238), and the mean gestational age was 37±3 weeks (range: 25–41 weeks; n=245). Premature delivery (<37 weeks of gestation) occurred in 35.5% (n=87/245) of patients, and SGA was found in 35.3% (n=83/235) of patients. The percentage of each disease type was as follows: MsMD: 69.0%; CM: 13.5%; LS: 9.3%; and HD: 8.2%. Overall, 44.8% of patients died. The mortality rate was significantly higher in patients with CM (60.5%) than in those with LS (23.1%) (p=0.03). There were significantly more early deaths within 2 days after birth in MsMD than in LS cases (p=0.03) and significantly more early onset within 2 days after birth than in HD cases (p=0.02).
Characteristics of each disease type in 281 newborns with neonatal-onset mitochondrial disease
Of the 203 patients for whom blood test results were obtained, 175 (86.2%) showed hyperlactacidaemia. MsMD cases had significantly more hyperlactacidaemia than CM cases (p=0.006).
The overall survival curve is shown in figure 2. The median survival time was 1.86 years, with a 95% CI of 0.90 to 2.95 years. The median survival rate at 1 year was 51.8% (95% CI 44.8% to 58.5%).
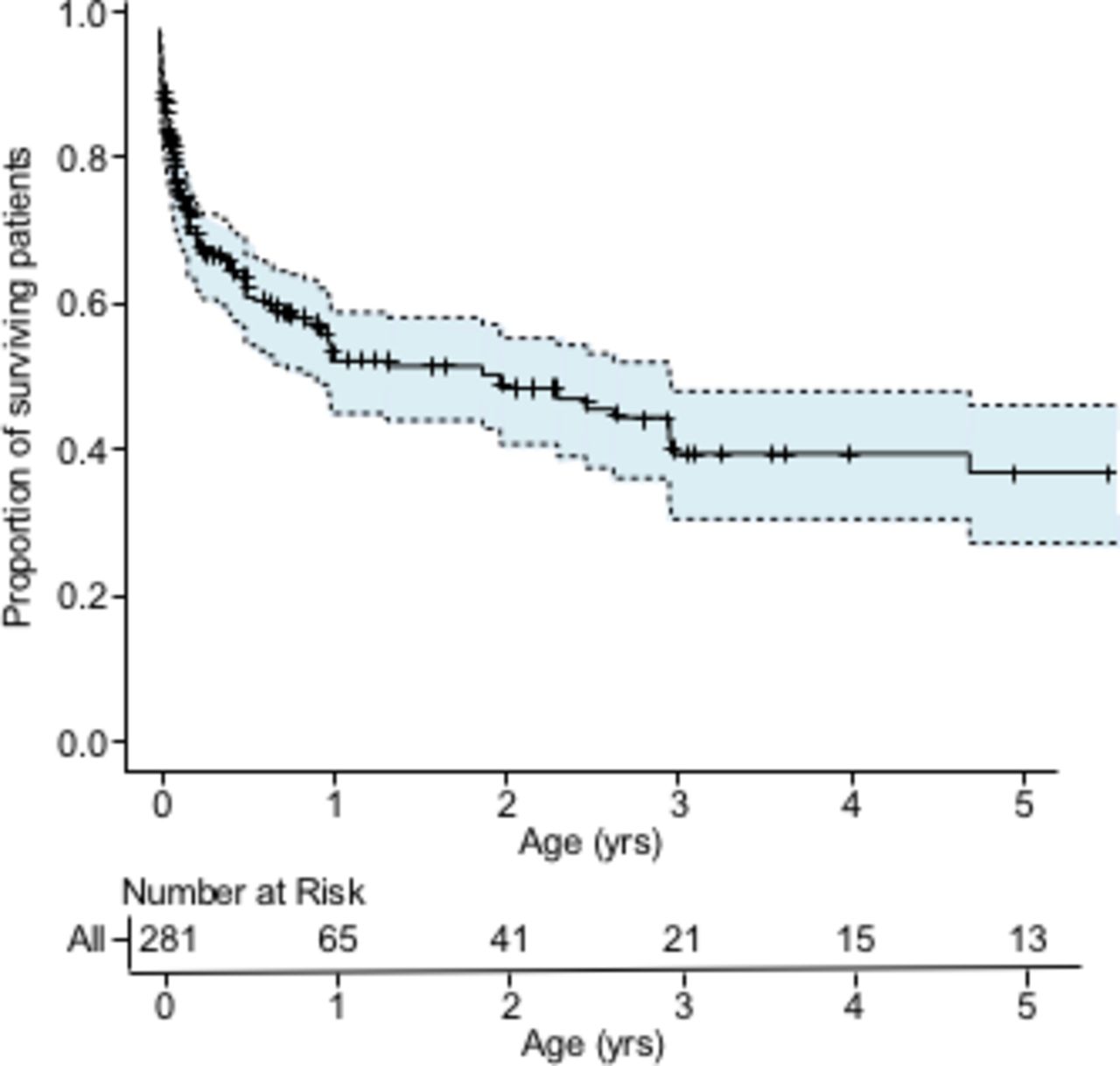
Kaplan-Meier analysis of overall survival with numbers of subjects at risk. The tick marks show censored cases. The shaded area indicates the 95% CI.
Initial symptoms are summarised in table 2. Patients presented with one or more of the initial symptoms. Onset within the first 2 days after birth was observed in 236 (73.5%) patients. Symptoms in the other 55 patients presented between 3 and 26 days of life (median: 4 days). The most common initial symptom was respiratory disorder (n=83, 25.9%), followed by neonatal asphyxia (n=46, 14.3%) and poor feeding or failure to thrive (n=34, 10.6%).
Initial symptoms by day of onset in 281 newborns with neonatal-onset mitochondrial disease (N*=321)
Molecular diagnosis
Of the 84 (29.9%) molecularly diagnosed patients, 69 (24.6%) had nuclear DNA variants in 36 genes (ACAD9, AIFM1, ATAD3, BOLA3, C1QBP, COQ4, COX10, DGUOK, EARS2, ECHS1, FBXL4, GFM1, GTPBP3, HSD17B10, IBA57, LRPPRC, MPV17, NDUFA10, NDUFAF6, NDUFB11, PC, PDHA1, PTCD3, QRSL1, RARS2, RMND1, SDHA, SERAC1, SLC19A3, SLC25A4, SLC25A26, SUCLA2, SUCLG1, TAZ, TMEM70 and TUFM). Among the other 15 (5.3%) patients, 11 had mitochondrial DNA variants in five genes and four had a single large deletion.
Among the 271 patients diagnosed using the biochemical approach, the molecular diagnosis was confirmed in 74 (27.3%). In patients with complex I, complex IV or combined deficiency, the molecular diagnosis was confirmed in 26.6%, 13.6% and 50.7%, respectively. There were no molecularly diagnosed patients of complex II and III deficiency. Of the 89 patients diagnosed by decreased OCR, 13 (14.6%) had a confirmed molecular diagnosis.
Prognostic factors
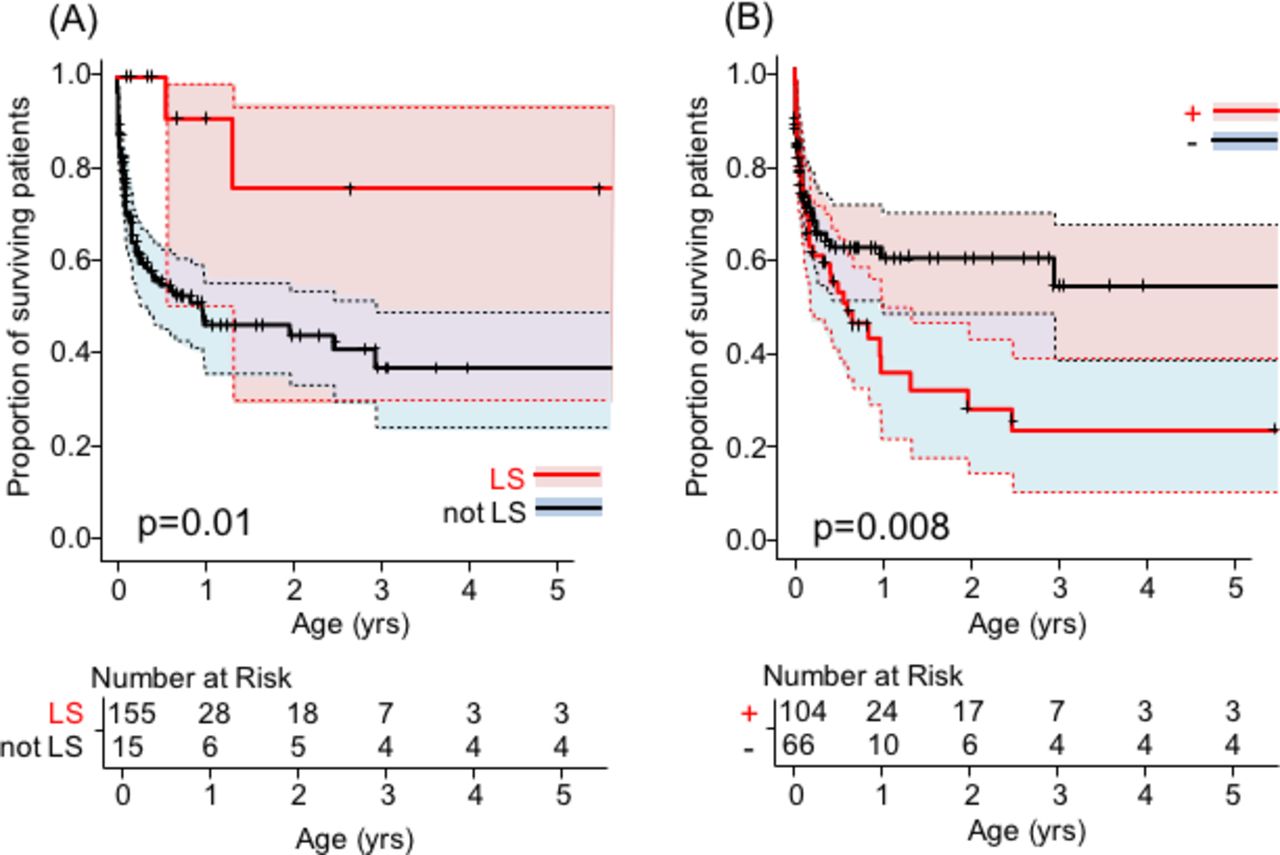
In the log-rank test, there was a significant difference in prognosis between patients with and without LS and molecular diagnosis (p=0.007 and p=0.03, respectively). The prognostic effect of hyperlactataemia was also suggested, but there was no significant difference (p=0.08). The Cox proportional hazards regression analysis of the effects of hyperlactacidaemia, molecular diagnosis and LS on the survival curve showed the independent and significant effects of LS (HR=0.15, 95% CI 0.04 to 0.63, p=0.01) and molecular diagnosis (HR=1.87, 95% CI 1.18 to 2.96, p=0.008) on survival. The two survival curves are shown in figure 3.
{kind=link}
{kind=link}
{kind=link}
Association of survival rates with significant factors. Multivariate analysis with Cox proportional hazards regression analyses. (A) Survival rate by the presence of Leigh syndrome (LS) with the number of subjects at risk. The red colour indicates patients with LS. The blue colour indicates patients without LS. Patients with LS had a significantly lower mortality rate (p=0.010). The tick marks show censored cases. (B) Survival rate by the presence of molecular diagnosis with the numbers of subjects at risk. The red colour indicates patients with molecular diagnoses. The blue colour indicates patients without molecular diagnoses. Patients with molecular diagnoses had a significantly lower mortality rate (p=0.008). The tick marks show censored cases. The shaded areas show the 95% CIs.
Discussion
Studies in a large number of patients with neonatal-onset MD are limited. In this study, we characterised several clinical features, molecular diagnosis and prognosis in a larger cohort of patients with neonatal-onset MD.
The frequency of both preterm birth and SGA was 35%, which was similar to the previously reported rates of 25%–30% and 22%–36%, respectively.1 2 Other reports have shown a relationship between MD and intrauterine growth restriction, but not between MD and preterm birth.18 19 Although it is difficult to determine whether neonatal-onset MD is a risk for preterm birth due to the different case backgrounds, intrauterine growth retardation was reconfirmed as one of the characteristics of neonatal MD.
As for the disease types, MsMD accounted for the majority (69%) of the cases because the exclusion criteria for the other types make it the default. However, the concept of MsMD as a disease is less established, and hence, clear diagnostic criteria are needed in the future. Relative to the overall population onset from infancy to adulthood,9 the proportion of LS decreased, and the proportion of CM increased.
The neonatal mortality rate was 23%, indicating that the prognosis is still poor compared with that reported previously (15%–41%).1 2 By disease type, mortality was higher in patients with CM (61%) and lower in those with LS (23%). A poor prognosis for CM has been reported previously.20 Mortality in the neonatal period was significantly higher for MsMD than for LS. The low all-cause mortality in LS is partially considered to be due to the low neonatal mortality rate.
Disease onset within the first 2 days of birth was 74%, with is higher than that in previous studies (51%–59%).1 2 This is thought to be due to the fact that MD is now being diagnosed when a patient’s general condition deteriorates immediately after birth and early death occurs. Neonatal asphyxia of an unknown cause and sudden respiratory distress are common immediately after birth, and poor feeding, failure to thrive, drowsiness or poor health condition are common 2 days after birth, as previously reported.1 In addition, MsMD presented an earlier onset than HD. The reason for the late onset of HD was thought to be that cholestasis (jaundice), one of the main symptoms of HD, is common 2 days after birth.
With respect to biochemical findings, hyperlactacidemia was observed in 86% of patients, as in previous reports (77%–87%).2 18 It should be noted that the frequency is lower in organ-specific disease types such as CM. OCR was used in the diagnosis of MD in 32% of patients. OCR measurement tends to be used as a screening tool in patients with clinically suspected MD.21 The combination of the MRC enzyme assay and the OCR measurement method improves the diagnosis rate of LS.12
With respect to molecular diagnoses, 30% patients had genetic variants, 81% of which were in nDNA. There are no recent large cohort reports of genetic variants in neonatal MD to compare with. The higher molecular diagnosis rate of combined complex deficiencies (51%) than that of a complex I and IV deficiency (27% and 14%) is thought to be due to the fact that many of the pathogenic genes are involved in other metabolic pathways rather than oxidative phosphorylation subunits and assembly factors.8 On an average, 22 novel, disease-related genes have been identified each year since the widespread adoption of next-generation sequencing (NGS).22 A comprehensive analysis of Japanese data targeting childhood-onset MD was reported in 2016 and showed that a causative gene could be identified in 35% of cases.5 Compared with overall childhood-onset MD, neonatal-onset MD still has a lower rate of molecular diagnosis. One reason for this is probably the difficulty in clinically diagnosing neonatal-onset MD, which presents with non-specific symptoms and is prone to developing into severe disease.
Among the patients diagnosed using the biochemical approach and with a confirmed molecular diagnosis, 18% (n=13/71) did not have decreased MRC enzyme activity and were screened for decreased OCR. The diagnosis of neonatal-onset MD is based on a general method for diagnosing MD,10 but making a diagnosis is challenging because many patients die early during the clinical course, and the collection of samples during survival is difficult. If necessary, a combined analysis of MRC enzyme assay and OCR measurement will help confirm the diagnosis of neonatal-onset MD, which is difficult to diagnose clinically. On the contrary, depending on local accessibility to broad NGS approaches, diagnostic algorithms for MDs tend to favour genetic first over phenotype first.13 As we observed in a small number of patients, certain patients with possible MD cannot be diagnosed using biochemical analyses alone23 but could be diagnosed molecularly. However, biochemical tests are still valuable in critically ill patients for whom time is an issue and for functional validation of class 3 variants to establish a firm diagnosis. It is important to perform comprehensive testing by collecting appropriate tissue samples, while considering invasiveness, and by analysing the genes responsible.
Both LS and molecular diagnosis had significant effects on prognosis. Although neonatal onset has been shown to be a poor prognostic factor for LS in childhood,24 it was found to be a good prognostic factor in the present study, where we compared only patients with neonatal onset. Against the possible influence of genetic background, a larger number of cases should be studied. The poor prognosis of patients with a molecular diagnosis suggests that patients with a mitochondrial dysfunction based on a biochemical analysis may include secondary mitochondrial dysfunctions other than MD. Therefore, prognosis should be separate for patients whose mitochondrial dysfunction is detected using biochemical analyses and those with a molecular diagnosis.
Finally, although this study was conducted in a larger group than that used in previous studies, the number of cases is still low due to the diversity of pathogenesis and genetic variations. Therefore, it is necessary to study more cases to validate the outcomes.
Conclusions
Neonatal-onset MDs show diverse pathogenesis and genetic aetiologies. The number of diagnoses can be increased, and clarity regarding prognosis can be achieved with comprehensive biochemical and molecular analyses using appropriate tissue samples. It is necessary to continue to study cases and, as genetic testing becomes more widespread and advanced, to use them in larger studies to clarify the pathogenesis of neonatal-onset MD.
Data availability statement
Data are available on reasonable request. Deidentified participant data are available from corresponding author, Kei Murayama. kmuraya@mri.biglobe.ne.jp. ORCID iD 0000-0002-3923-8636. Data are available on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study was approved by the Institutional Review Board in Saitama Medical University (numbers 482 and 844).This study was approved by the ethics boards of the Chiba Children’s Hospital, Saitama Medical University, and Juntendo University.
Acknowledgments
The authors would like to thank all the patients and their physicians for allowing us to analyse their data.
References
Footnotes
Contributors TE, TN and KM conceptualised and designed the study, drafted the manuscript, collected the data, carried out the initial analyses and reviewed and revised the manuscript. YS, TT, KI, AM, NA, MO-T, YoO, MS, MT, TM, YY, KRN, YK, TF, AtO, AkO and YaO coordinated and supervised data collection and critically reviewed the manuscript for important intellectual content.
Funding This work was partially supported by the Practical Research Project for Rare/Intractable Diseases from the Japan Agency for Medical Research and Development, AMED (JP21ek0109468, JP19ek0109273, JP21kk0305015.)
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.