Article Text

Abstract
Objective To characterise the phenotype and genotype of neonates born small-for-gestational age (SGA; birth weight <10th centile) who developed hyperinsulinaemic hypoglycaemia (HH).
Methods Clinical information was prospectively collected on 27 SGA neonates with HH, followed by sequencing of KCNJ11 and ABCC8.
Results There was no correlation between the maximum glucose requirement and serum insulin levels. Serum insulin level was undetectable in five infants (19%) during hypoglycaemia. Six infants (22%) required diazoxide treatment >6 months. Normoglycaemia on diazoxide <5 mg/kg/day was a safe predictor of resolved HH. Sequencing of KCNJ11/ABCC8 did not identify any mutations.
Conclusions Serum insulin levels during hypoglycaemia taken in isolation can miss the diagnosis of HH. SGA infants may continue to have hypofattyacidaemic hypoketotic HH beyond the first few weeks of life. Recognition and treatment of this group of patients are important and may have important implications for neurodevelopmental outcome of these patients.
- Hyperinsulinism
- Hypoglycaemia
- Small for Gestational Age Infant
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
What is already known on this topic
-
Infants born small-for-gestational age are at an increased risk to develop transient hyperinsulinaemic hypoglycaemia (HH).
-
Mutations in ABCC8/KCNJ11 genes are the commonest cause of congenital HH.
What this study adds
-
This largest series of small-for-gestational age infants who developed hyperinsulinaemic hypoglycaemia (HH) highlights that hypofattyacidaemic hypoketotic HH may persist beyond the first few weeks of life.
-
HH in these infants is diazoxide responsive and not identified to be due to mutations in ABCC8/KCNJ11 genes in this series.
-
Withdrawal of diazoxide treatment when glycaemic control is maintained at a dose of <5 mg/kg/day can be safely recommended.
Introduction
Hyperinsulinaemic hypoglycaemia (HH) is characterised by the unregulated secretion of insulin from pancreatic β-cells in relation to the blood glucose concentration. It is the commonest cause of severe and persistent hypoglycaemia in the neonatal and infancy periods.1 The genetic basis of congenital forms of HH involves defects in key genes (commonest—ABCC8 and KCNJ11 encoding the two subunits SUR1 and Kir6.2, respectively), which regulate insulin secretion from pancreatic β-cells.1 HH may be secondary to certain risk factors (such as maternal diabetes mellitus, perinatal asphyxia and small-for-gestational age (SGA)). In 1984, Collins and Leonard first described transient HH in SGA infants.2 It has not previously been documented whether the HH observed in infants born SGA is due to defects in the ABCC8/KCNJ11 genes. The phenotype of a large cohort of SGA neonates who developed HH has not been studied previously. The purpose of the present study was to report on the phenotype, clinical course and results of the sequencing of ABCC8/KCNJ11 in a cohort of infants born SGA who developed HH.
Patients and methods
We prospectively studied the clinical and biochemical features of 27 SGA (defined as birth weight <10th centile) neonates with HH who were admitted to two tertiary paediatric referral centres (Great Ormond Street Hospital and Bristol Royal Hospital for Children). Neonates with history of perinatal asphyxia, Rhesus isoimmunisation and syndromic forms such as Beckwith-Wiedemann syndrome were excluded from the study. All were investigated at the time of clinical and biochemical hypoglycaemia to establish the diagnosis of HH and were commenced on diazoxide (5–20 mg/kg/day). Diazoxide responsiveness was defined as ability to come off intravenous glucose and maintain normoglycaemia during a physiological fast. Following discharge, they were followed up at 3-monthly intervals in outpatient clinic, where glycaemic control and diazoxide dose were reviewed. These infants were readmitted for a 24-h glucose profile and a controlled fast after stopping diazoxide for 3 days, if home glucose monitoring indicated no recurrence of hypoglycaemia on <5 mg/kg/day of diazoxide.1 If home glucose monitoring revealed hypoglycaemic episodes, the dose of diazoxide was adjusted and the trial off diazoxide was postponed until the dose was <5 mg/kg/day.
Genetics
Genomic DNA was extracted from peripheral leukocytes using standard procedures and the coding regions and intron/exon boundaries of the ABCC8 and KCNJ11 genes were amplified by PCR (primers available on request). Amplicons were subsequently sequenced using the Big Dye Terminator Cycler Sequencing Kit V.3.1 (Applied Biosystems, Warrington, UK) according to manufacturer's instruction and reactions were analysed on an ABI 3730 Capillary sequencer (Applied Biosystems, Warrington, UK). Sequences were compared with the reference sequences (NM_000525.3 and NM_000352.3) using Mutation Surveyor v3.24 software (SoftGenetics, State College, Pennsylvania, USA).
Results
Clinical characteristics and course
The phenotypic details of 27 SGA infants with HH are summarised in table 1.
Clinical characteristics of 27 small-for-gestational age infants with hyperinsulinaemic hypoglycaemia
The plasma insulin levels were inappropriately elevated during hypoglycaemia in 22 infants (81%). The remaining five infants (19%) had inappropriately low β-hydroxybutyrate and non-esterified fatty acids during hypoglycaemia as supportive biochemical evidence of HH. All infants required glucose infusion rate of ≥8 mg/kg/min to maintain normoglycaemia (median 15.6 mg/kg/min). There was no correlation found between maximum glucose infusion rate to maintain normoglycaemia and plasma insulin level during hypoglycaemia (figure 1). Most (89%) responded to mild–moderate doses of diazoxide (5–10 mg/kg/day), with three infants (11%) needing high doses (15–20 mg/kg/day).

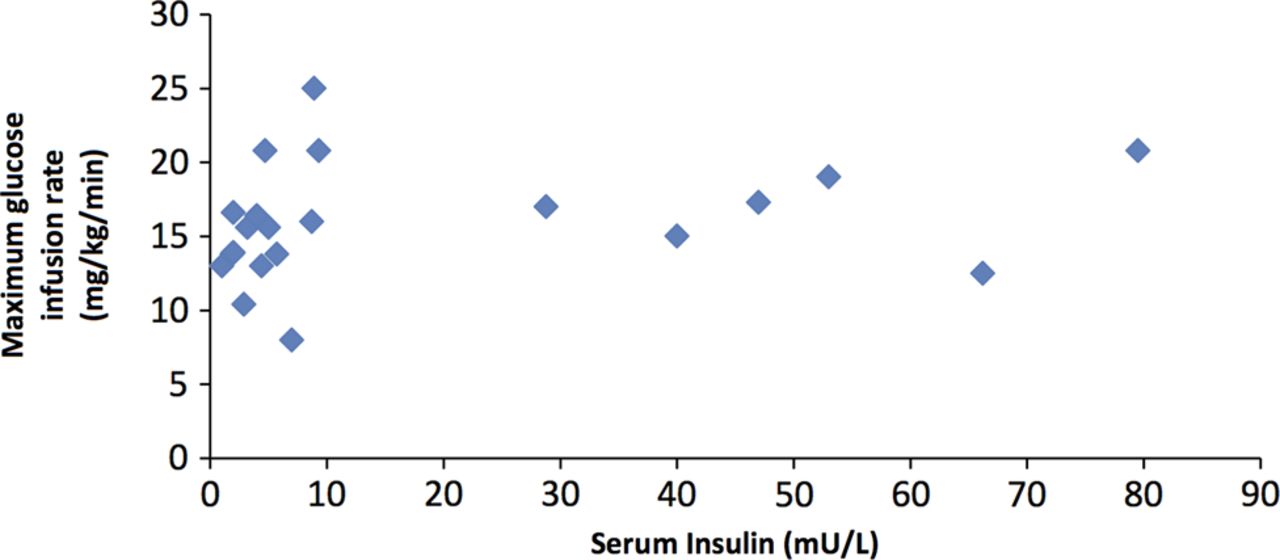{kind=link}
Figure demonstrating the lack of correlation between maximum glucose infusion rate and plasma insulin concentrations.
Glucose profile and fasting responses at resolution of hyperinsulinism
In all, 17 infants (63%) underwent a 24 h glucose profile and controlled fast after stopping diazoxide to demonstrate resolution of hyperinsulinism. None of these infants recorded hypoglycaemia during a 24-h glucose profile. All infants maintained blood glucose concentrations above 3.0 mmol/l at the end of fast appropriate for their age, with 13 infants maintaining blood glucose concentrations >3.5 mmol/l. All infants showed appropriate plasma insulin suppression and elevation of plasma β-hydroxybutyrate/non-esterified fatty acids as evidence of resolution of HH.
Genetic results
Sequencing did not reveal mutations in ABCC8/KCNJ11 genes in 25/27 DNA samples analysed.
Discussion
This is the largest case series of SGA neonates with HH that reports the clinical characteristics and ABCC8/KCNJ11 sequencing results in a cohort of 27 subjects. Collins et al first described transient HH in three SGA infants and subsequently reported HH in five of 27 SGA neonates studied, suggesting it is common in SGA neonates.2 ,3 It is important to recognise this as neonates with HH are especially prone to the complications of hypoglycaemia because of their inability to generate alternative fuels, such as ketone bodies.
In our study, despite other evidence of hyperinsulinism, plasma insulin levels were undetectable during hypoglycaemia in 5/27 SGA neonates. We did not find any correlation between the maximum glucose infusion rate required to maintain normoglycaemia and plasma insulin levels. This is explained by the short half-life of insulin of 4–5 min and rapid clearance by the liver before it reaches peripheral circulation. In a study by Hoe et al,4 plasma insulin was inappropriately elevated in only 11 of 24 neonates who had transient prolonged neonatal hyperinsulinism. Our results corroborate these findings and highlight the value of other parameters (apart from plasma insulin) in the diagnosis of HH.
In this case study of SGA infants referred to two tertiary hospitals with HH, all infants responded to diazoxide, though the dose was variable ranging from 5 to 20 mg/kg/day. Diazoxide was successfully stopped in 21 (78%) patients by 6 months of age. In the remaining six infants, five needed diazoxide until 10 months of age and one infant needed prolonged treatment until 22 months. In a study of 26 neonates with hyperinsulinism which included 7 SGA neonates, HH resolved by a median age of 181 days (range, 18–403 days).4 Fafoula et al5 also reported prolonged transient HH up to 9 months in SGA infants.
In our cohort, no recurrence of HH was noticed after stopping diazoxide. This highlights that with the management protocol whereby diazoxide was stopped when good glycaemic control was maintained with diazoxide dose <5 mg/kg/day, the risk of recurrence of hypoketotic hypoglycaemia brain injury to these vulnerable infants is avoided.
To our knowledge, no previous study has reported on the genetic results of SGA neonates who developed HH. In our study, no mutations in ABCC8/KCNJ11 were identified in these SGA infants. However, rarer genetic causes of HH due to mutations in GLUD1, HADH, GCK and HNF4A were not excluded.
Conclusions
SGA infants may continue to have hypofattyacidaemic hypoketotic HH beyond the first few weeks of life. Recognition and treatment of this group of patients are important and may have important implications for neurodevelopmental outcome of these patients. Plasma insulin levels during hypoglycaemia taken in isolation can miss the diagnosis of HH and hence other parameters (such as glucose infusion rates and production of free fatty acids/ketone bodies during hypoglycaemia) must be considered in the diagnosis. Withdrawal of diazoxide treatment when glycaemic control is maintained at a dose of <5 mg/kg/day can be safely recommended.
Finally, the genetic aetiology of HH in SGA infants is not understood and, in this study, mutations in the common genes implicated in the aetiology of congenital hyperinsulinism were not identified. Further studies are required to understand the underlying mechanism of HH in these infants.
Acknowledgments
This study was funded by the Wellcome Trust (081188/A/06/Z). VBA was funded by MRC, SEF was the Sir Graham Wilkins Peninsula Medical School Research Fellow, SE was funded by the Royal Devon & Exeter NHS Foundation Trust Research & Development Directorate and RRK was funded by NIHR.
Footnotes
-
Contributors VBA collected and analysed the data and wrote the manuscript. SEF performed the genetic studies and reviewed the manuscript. AK and JPS collected the data and critically reviewed the manuscript. SE and KH conceptualised the study and critically reviewed the manuscript. RRK conceptualised the study, analysed the data and critically reviewed the manuscript. All authors contributed to the approval of the final version of the manuscript. RRK is the guarantor of this work.
-
Funding This work was funded by the Wellcome Trust (081188/A/06/Z).
-
Competing interests None.
-
Ethics approval Ethics Committee of Great Ormond Street Hospital for Children and the Institute of Child Health, University College London.
-
Provenance and peer review Not commissioned; externally peer reviewed.