Article Text

Abstract
Background Less-invasive surfactant administration (LISA) is increasingly used. We investigated the feasibility of a new LISA-device (Neofact®) in neonates.
Design Prospective observational pilot study with open-label LISA in two tertiary neonatal intensive care units.
Patients 20 infants with a gestational age of ≥26+0/7 weeks and an indication for LISA (Respiratory Severity Score (RSS)≥5 or fraction of inspired oxygen (FiO2) ≥0.30). Infants with respiratory tract malformations or unavailability of an instructed neonatologist were excluded.
Main outcome measures Success of LISA, defined as laryngoscopy-confirmed intratracheal catheter position or a decrease in FiO2 by ≥0.05 or to 0.21, accompanied by an RSS decrease of ≥2; number of attempts needed for tracheal catheterisation.
Results 20/57 screened infants were enrolled. Successful application occurred in 19/20 (95%). One application failed after three attempts. No device-related adverse events occurred. The median number of attempts was 2, success rate per attempt 19/31 (61%).
Conclusion LISA via Neofact® appears feasible.
- neonatology
- resuscitation
- technology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
What is already known on this topic?
Treatment of respiratory distress syndrome (RDS) in neonates via less-invasive surfactant administration (LISA) is quite common in Europe and in some other countries.
The surfactant application device Neofact® was developed to perform LISA in neonates and is now approved as a medical device in the European Union.
What this study adds?
This study evaluated the clinical feasibility of the new device in neonates with RDS and a gestational age of ≥26+0/7 weeks.
Additionally, an instructional video has been produced and added as online supplemental material.
Supplementary video
Background
Less-invasive surfactant administration (LISA) to spontaneously breathing neonates is common in Europe.1 LISA requires advancing a thin catheter into the trachea, mostly using a Magill forceps for soft catheters.2 We developed an application device (Neofact®) rendering this forceps, tested it on a manikin3 and now set out to monitor its clinical feasibility. Secondary objectives were procedure and laryngoscopy duration, handling time of the device and the number of attempts needed for inserting the catheter intratracheally. Additionally, any need for secondary intubation/mechanical ventilation (MV) and occurrence of complications were determined.
Methods
This is a bicentric, one-arm, prospective observational study of a CE-marked medical device.
Population
A convenience sample of 20 preterm infants with respiratory distress syndrome (RDS) and a fraction of inspired oxygen (FiO2) of ≥0.30 or a modified Silverman-Andersen Respiratory Severity Score (RSS) of ≥54 while receiving non-invasive respiratory support were studied. Exclusion criteria were a gestational age (GA) of <26+0 weeks, any respiratory tract malformations, a clinical decision to apply surfactant via an endotracheal tube or an attending physician not trained by the principal investigator.
Device
The angles of the device correspond to a Magill forceps, and care was taken to construct a soft tip to avoid injuries. The plastic components have Food and Drug Administration and United States Pharmacopeia approval for medical devices. The tunnel has a length of ~90 mm and a cross section of 4 mm. The catheter is soft, with 3.5 Fr diameter, 550 mm length, 0.2 mL dead space and a black marked tip (10 mm, figure 1).

Surfactant application device (Neofact®).
Intervention
Device insertion was controlled via video laryngoscopy; the latter being removed once the catheter had been inserted. Investigators were manikin-trained prior to enrolment. No more than three attempts were allowed; in case of complications, investigators could stop the application at any time, in which case LISA was carried out as per local standard (nasally guided catheter). Following LISA, infants were observed for 48 hours to document potentially device-related adverse events, additional surfactant doses and need for MV. Alveofact (45 mg/mL; Lyomark, Oberhaching, Germany) was used at 100 mg/kg.
Outcome measures
Success of the procedure was defined as endoscopically verified intratracheal catheter position or clinical response (decrease in FiO2 by ≥0.05 or to 0.21 accompanied by a ≥2-point RSS decrease within 30 min).
Procedure duration was defined as the time from initial laryngoscope insertion to device removal, laryngoscopy duration as the time the laryngoscope blade was in the patient’s mouth and handling time from onset of device insertion to laryngoscope removal. The total duration of laryngoscopy was the sum of all attempts. Time intervals and the number of attempts needed for catheterisation were derived from the video-laryngoscope recordings with an accuracy of ±1 s using VLC media player V.3.0.8.
Complications, including onset and severity of coughing, choking, apnoea and laryngospasm, were documented by the physician in charge. A second person documented the nadir pulse oximeter saturation (SpO2), number of desaturations (SpO2 <80%), bradycardia (<80 beats/min), tachycardia (>200 beats/min), arterial hypotension (mean arterial pressure (MAP)<GA) or hypertension (MAP>GA +20 mm Hg) during the procedure.
Implementation
Infants were recruited during delivery room management or postnatal hospitalisation. Blinding was not feasible due to the nature of the intervention.
Statistics
Analysis was performed using SAS V.9.4. Endpoints were evaluated by calculating percentages and 95% CIs. Continuous outcomes are given as mean±SD or median with IQRs or minimum/maximum. All evaluations are purely descriptive.
Safety
The study was monitored by the local Centre for Paediatric Clinical Studies according to the guidelines on good clinical practice. A steering committee monitored procedure safety after every five inclusions.
Results
Recruitment and population
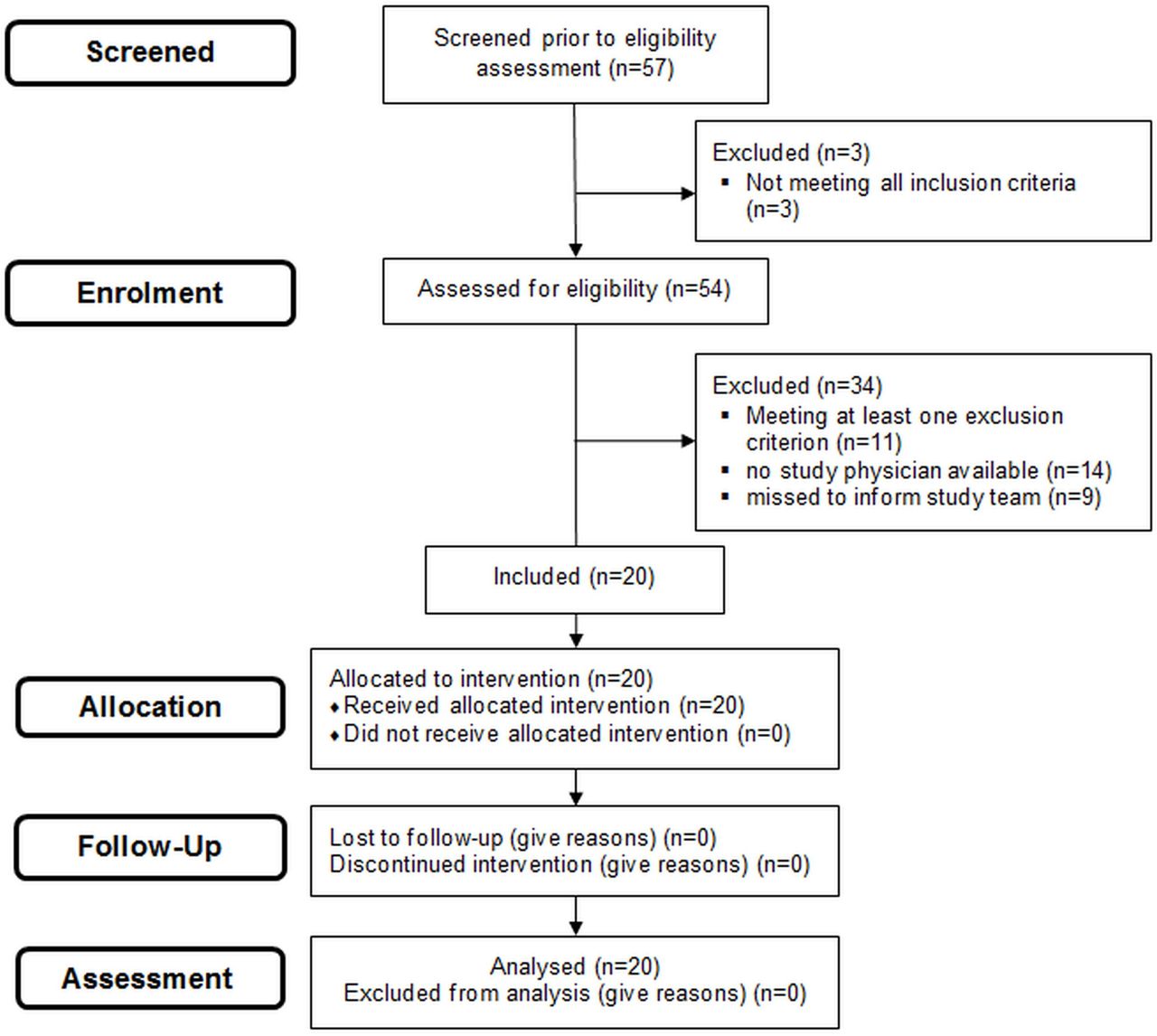
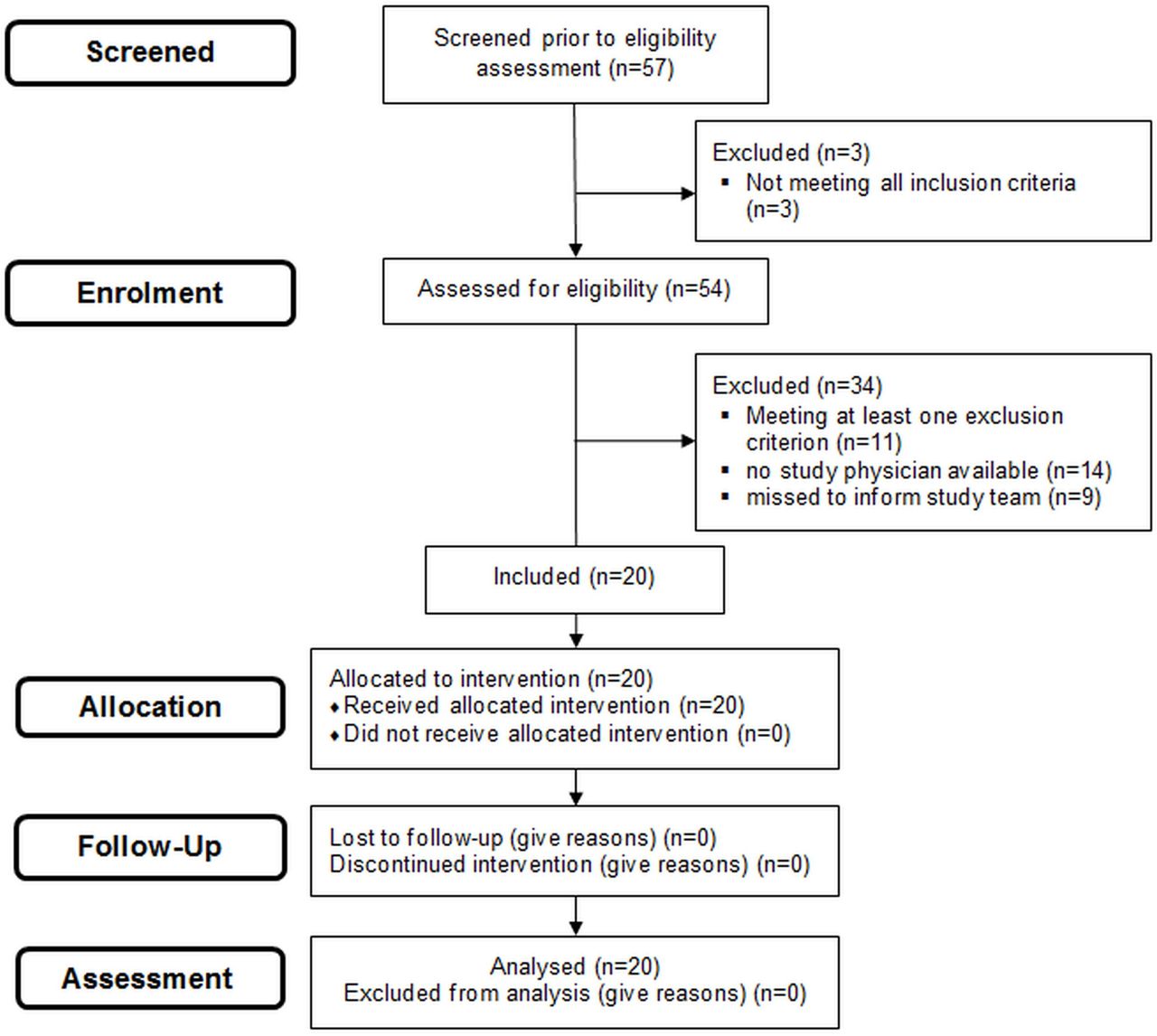
For the study flow see figure 2.
{kind=link}
{kind=link}
Patient flow.
Recruitment took place between May 2019 and October 2019. Each centre recruited 10 consecutive infants (35%) at a mean±SD age of 12±8.8 hours (range: 24 min–25 hours). The mean±SD GA of included infants was 315/7±32/7 weeks and the birth weight was 1691±704 g. Three interventions took place in the delivery room, and all 20 infants were diagnosed with RDS. Baseline characteristics of included infants and the number of analysed infants/videos for each outcome criterion are shown in table 1.
Baseline characteristics of included infants/number of analysed videos for each outcome criterion
Outcome
19 of 20 procedures showed success (95%, 19 laryngoscopy-verified, 16 infants responded clinically). One application was stopped after three attempts. Success rate per attempt was 19/31 (61%; three failed attempts were laryngoscopy-related, ie, terminated prior to device insertion, and thus were excluded).
The median duration of the entire LISA procedure and laryngoscopy of successful attempts was 499 (IQR 373–758) s and 79 (IQR 62–87) s, respectively; handling time of the device was 42 (IQR 29–64) s.
Before intervention, 8 infants underwent continuous positive airway pressure (CPAP) and 12 synchronised nasal intermittent positive pressure ventilation (SNIPPV); afterwards, 2 infants received no respiratory support, 2 CPAP, 11 SNIPPV and 2 MV.
Ten infants received additional surfactant doses via the standard LISA procedure (if not intubated).
Complications
Two infants developed stronger choking; none were coughing heavily. One infant showed laryngospasm, but catheter insertion was successful on a second attempt. One apnoea led to stopping the attempt. Mean nadir SpO2 was 59%±16%. There were no device-related adverse events (including mucosal erosion).
Ancillary analyses
Sixteen infants received sedatives (propofol, morphine or midazolam). Heart rate and MAP remained largely unchanged.
Discussion
We set out to evaluate feasibility of our device in neonates and were encouraged by a high success rate (95%). However, the intervention was performed by LISA-experienced neonatologists who still required a median of two attempts to succeed. This might be due to the novel procedure (none had used the device before in infants) or the study settings:
LISA was performed via video laryngoscopy, with which most investigators were less experienced (also reflected by the ratio of laryngoscopy to handling time and a high rate of difficulties apparent in the video recordings). Success may increase with experience, but this remains speculative. Since the device is modelled to a Magill forceps, absence of a learning curve is considered unlikely.
The study was conducted in neonates with a mean GA of >30 weeks, which reflects not the ‘typical’ LISA population. We accepted this and a higher rate of laryngoscopy attempts5 due to the device’s first use in infants, which also explains why procedure time was longer than in our manikin study. This might also explain the higher rate of sedative use and apnoeas, as older infants are more likely to fight laryngoscopy, limiting the primary endpoint.
Taking this into account, first attempt success rate was regarded comparable to published data.5 6
All secondary outcomes were evaluated descriptively. However, results were considered in agreement with common experience in this population. A comparison with previous LISA procedures would be desirable but appeared unfeasible with the data available.
Our physician selection might have introduced bias but was indispensable due to the need for prior instructions. The manufacturer sponsored the study, and the first author invented the device. However, all examinations were conducted by physicians with no financial relationship to either.
Conclusion
LISA via Neofact appears feasible. Complications seemed to be rare. Further research is needed to determine the feasibility in neonates with a GA of <26 weeks.
Acknowledgments
The Neofact feasibility study investigators acknowledge the contribution of Ingolf Grahl, Paul Seidel, Franziska Trautmann, Cornelia Hoelzl and their team at the Murrplastik Medizintechnik GmbH (Falkenstein, Germany) as product producers and engineers of the device; and Christian Kuhring, Niklas Nagel, Malik Malocho and Christian Parvany and their team at the Lyomark Pharma GmbH (Oberhaching, Germany) as distribution partners of the device and supporters in the equipment and financing of the study.
Footnotes
Collaborators Neofact® - feasibility Study Group: Coordinating Investigator: Christian A. Maiwald (University Children’s Hospital Tübingen, Department of Neonatology, Tübingen, Germany; corresponding author). Steering Committee: Axel R. Franz, Corinna Engel and Christian F. Poets (University Children’s Hospital Tübingen, Department of Neonatology, Tübingen, Germany); Patrick Neuberger and Matthias Vochem (Olgahospital Stuttgart, Department of Neonatology, Stuttgart, Germany). Data Management, Biometry, Monitoring and Study Coordination: Iris Bergmann, Corinna Engel, Andrea Evers-Bischoff (all at the Center for Pediatric Clinical Studies, University Children’s Hospital Tübingen, Germany). Recruiting Hospitals and Local Investigators: Olgahospital Stuttgart, Department of Neonatology: Patrick Neuberger and Christian Schlunk. University Children’s Hospital Tübingen, Department of Neonatology: Axel R. Franz, Christian Gille, Ingo Müller-Hansen, Jörg Arand, Kerstin Gründler, Karen B. Kreutzer and Nicole Wolf.
Contributors CAM evolved the study protocol under the supervision of AF, PN, MV, CE and CFP;
supervised and gave helpful annotations on request in some of the surfactant administrations in Tuebingen; and drafted the first version of the manuscript. AF and PN acted as local principal investigators. CAM and the local investigators of the study group informed parents and obtained their consent for participation. Surfactant administration was accomplished by all local investigators (excepting CAM). CE and CAM analysed the data. AF, CFP, PN and MV reviewed the manuscript with respect to clinical interpretation of the data. All authors reviewed the manuscript, contributed to each draft version and approved the final version of the manuscript.
Funding This research project was enabled by a research funding from the Lyomark Pharma GmbH. Lyomark Pharma GmbH had no influence in the preparation of data or the manuscript.
Competing interests CAM has a financial relationship in the profits of the product as an inventor. The other authors have indicated they have no financial relationships relevant to this article to disclose.
Patient consent for publication Not required.
Ethics approval This trial was performed in accordance with the Declaration of Helsinki and the guidelines of Good Clinical Practice. The study was evaluated and approved by the ethics committees of Tuebingen University Hospital (094/2018BO1) and the Medical Association of Baden-Württemberg (B-F-2018–107). Both waived consent for observation and administration of surfactant due to the device’s CE mark and the urgent indication for LISA; parental written informed consent was subsequently obtained for data recording and analysis.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.