Article Text

Statistics from Altmetric.com
Hypernatraemia is more often caused by water deficiency than by sodium excess. Water deficiency may be due to inadequate intake, abnormal losses or a combination of the two. Causes of hypernatraemia are listed in table 1.
Causes of hypernatraemia
HYPERNATRAEMIA CAUSED BY DEFICIENT WATER INTAKE
A mentally competent, healthy adult or child will not become hypernatraemic from water deficiency alone provided there is access to water. Even mild hypertonicity is a powerful stimulus to thirst. Hypernatraemia caused by inadequate intake is therefore confined to infants, the old and frail, and the mentally impaired except for the unusual circumstance that an individual has no access to water over a sustained period.
Several recent reports have described infants who became hypernatraemic in the neonatal period because of inadequate breast feeding by inexperienced mothers.1–3 This is not an argument against breast feeding, but makes the point that mothers need adequate support to establish lactation, especially for their first born infants, and that even though human milk has a low sodium concentration a minimum water intake is necessary to prevent dehydration and hypernatraemia.
HYPERNATRAEMIA CAUSED BY INCREASED RENAL WATER LOSS
Diseases that impair the ability of the kidney to concentrate urine lead to hypernatraemia if replacement of urinary water is inadequate. Provided the thirst mechanism is intact, hypernatraemia does not occur in the competent subject with access to water; polyuria and polydipsia are the cardinal symptoms in such cases. Young infants with severe concentrating defects are at high risk of hypernatraemia however, because the distressed crying caused by thirst is frequently misinterpreted. Central (cranial or hypothalamic) and nephrogenic diabetes insipidus (DI) are the best known causes of failure of urinary concentration, and typically present in infancy with recurrent episodes of dehydration with hypernatraemia.
Central DI is due to failure of synthesis or secretion of antidiuretic hormone (ADH). It is uncommon in infancy and usually secondary to acquired disease of or damage to the anterior hypothalamus,4 although a very rare, usually dominantly inherited, genetic form has been described.5 In older children it may be caused by Langerhans cell histiocytosis, hypothalamic or pituitary tumours such as craniopharyngioma, damage caused by encephalitis or meningitis, or infarction of the pituitary stalk (Sheehan’s syndrome).6 It is diagnosed by a carefully conducted water deprivation test. Treatment is by replacement therapy with 1-deamino(8-d-arginine) vasopressin (DDAVP, Desmopressin).
Nephrogenic DI (a poor term but one we are stuck with for historical reasons) is caused by resistance of the renal tubule to the action of ADH. ADH acts by binding to receptors on the distal tubule and collecting duct (V2 receptors), which leads to the insertion of water channels (aquaporin 2) in the apical (luminal) membrane of the tubule rendering it permeable to water. Two hereditary forms of nephrogenic DI exist: a rare X-linked recessive type caused by mutations in the gene for the V2 receptor7; and an even rarer autosomal recessive form caused by mutations in the gene for aquaporin 2.8 They are clinically indistinguishable except that the X-linked type affects only male infants. Neither form responds to exogenous ADH or DDAVP. Treatment is difficult. The aim in infancy is to supplement water intake to a level sufficient to achieve normonatraemia. Older children will generally maintain water intake in response to thirst.
Polyuria can be ameliorated by two strategies. The first is to give a low solute diet, since urinary solute excretion is a major determinant of urine flow rate. The diet should contain sufficient sodium, potassium and protein for normal growth with as little excess as is practicable. The second is to use a thiazide diuretic, which acts by producing mild extracellular fluid (ECF) volume contraction. This leads to increased reabsorption of filtered salt and water in proximal parts of the renal tubule, reducing the delivery of both to the diluting segment and hence reducing urine flow rate. Chronic administration of thiazides must be accompanied by potassium supplementation to prevent potassium depletion, or by the co-administration of a potassium retaining diuretic such as amiloride, which may further augment the effect of the thiazide in diminishing polyuria and polydipsia.9 A few reports have also appeared of a beneficial effect of non-steroidal anti-inflammatory drugs (NSAIDs), either alone or with a thiazide.10 The effect of NSAIDs is probably due to altered intrarenal haemodynamics, again with an increase in fractional reabsorption of filtered solute and water in proximal nephron segments.
Acquired renal diseases that damage the renal medulla can cause a form of “nephrogenic” (that is, vasopressin resistant) DI. These include sickle cell disease, reflux nephropathy and juvenile nephronophthisis (medullary cystic disease). The clinical features are similar to those of inherited nephrogenic DI (polyuria and polydipsia) but generally much less severe. Advanced chronic renal failure impairs the ability of the kidney both to dilute and to concentrate the urine, commonly resulting in mild polyuria and polydipsia.
Essential hypernatraemia is an uncommon condition that can be mistaken for central DI but has features that differentiate it from the latter. It occurs both in children and in adults. The features are recurrent episodes of hypernatraemia without obvious polyuria or thirst. All cases so far described have had hypothalamic damage or tumours. The condition appears to be caused by selective destruction of the osmoreceptor cells in the anterior hypothalamus, which leads to absence of thirst and ADH release in response to hypertonicity.11,12 In contrast to DI, baroreceptor mediated ADH release is intact, so that when the ECF becomes sufficiently contracted ADH is secreted irrespective of the tonicity of the body fluids at the time. Patients with this bizarre condition seem remarkably well most of the time, even though their plasma sodium concentrations may be greatly raised above normal. Treatment is with small doses of DDAVP which prevents or attenuates the hypernatraemia, which is presumably desirable in order to prevent further brain damage from recurrent hypertonicity.13
HYPERNATRAEMIA CAUSED BY NON-RENAL WATER LOSS
Increased evaporative water loss
The person lost in the desert, where evaporative water loss may be very high, can become hypernatraemic quite quickly if replacement of insensible loss is inadequate. A study from Israel14 demonstrated that young, fit men became significantly hypernatraemic after a few hours of desert marching even though they were provided with adequate water, and that they needed to be actively encouraged to drink if this was to be prevented. Another case report described a woman who became lost in the vicinity of Sodom and became severely hypernatraemic and encephalopathic.15 As pointed out in an early landmark review of hypernatraemia,16 this is strikingly reminiscent of a historical account of another woman in exactly the same locality who became so dehydrated that her body sodium and chloride apparently exceeded their maximum solubility concentration (Genesis 19: 26). The shipwrecked mariner in a lifeboat in the ocean is in an even worse predicament, since the salinity of sea water is such that if drunk it will inevitably raise the ECF sodium concentration even further than dehydration alone.
Hypernatraemic (hypertonic) dehydration
This is defined as dehydration with a plasma sodium concentration above 150 mmol/l, and usually caused by gastroenteritis. Although sodium and chloride are normally almost confined to the ECF, hypertonicity causes osmotic extraction of water from the cells so that there is in fact contraction of both major body water compartments. Calculations of estimated water loss based on ECF volume (typically about 25% body weight) therefore significantly underestimate the total deficiency, and such calculations should be based on total body water (60–70% of body weight depending on age—the great majority of cases occur in infancy). There has been a notable fall in the prevalence of hypertonic dehydration in the last three decades, at least in developed countries.17 This is because of changes in the composition of infant formula from skimmed cow’s milk to a more “humanised” profile, in particular lower total solute content. It is a common error to assume that it is the high salt content of cow’s milk that is to blame, but this is incorrect—even raw cow’s milk only has a sodium concentration of about 25 mmol/l, which is less than the concentration in fluids usually recommended for the treatment of hypertonic dehydration. The true explanation lies in the relatively high concentration of organic solutes (carbohydrate and protein).18,19 These are poorly absorbed by the small intestine of a child with gastroenteritis, and a large proportion of the ingested load enters the colon, where bacterial fermentation to smaller molecules increases the osmolality of the colonic fluid and leads to osmotic extraction of water from the circulation, thus raising the tonicity of the plasma. The products of fermentation are acidic, leading to titration of bicarbonate in the intestinal fluid and progressive acidosis, a consistent feature of hypertonic dehydration of this type.
Hypernatraemic dehydration can be effectively treated by oral administration of glucose/polyelectrolyte solution (for example, World Health Organization/Unicef rehydration solution), if tolerated, and this appears to be safer than intravenous rehydration.20 In the child with circulatory compromise, the circulation should first be restored with intravenous isotonic fluid, followed by slow correction of hypernatraemia with hypotonic saline (for example, 0.45% or half isotonic saline). This should be given at a rate calculated to lower the plasma sodium concentration by not more than 10–12 mmol/l per 24 hours. The reason for this is that, if hypertonicity has been present for more than a few hours, brain cells accumulate organic osmolytes (“idiogenic osmoles”) to maintain cell volume, and these are slow to dissipate.21 This predisposes to cerebral oedema if ECF tonicity is lowered too quickly.
Hypernatraemia caused by sodium excess
Iatrogenic hypernatraemia may result from the administration of repeated doses of hypertonic sodium bicarbonate to correct acidosis—for example, in the course of cardiopulmonary resuscitation. The inadvertent or misguided parenteral infusion of hypertonic saline (for which it is difficult to think of a legitimate indication) would have the same effect and has been described. As previously mentioned, iatrogenic hypernatraemia is more commonly the result of giving insufficient water than of sodium excess.
Accidental or unintentional salt poisoning has been described as a consequence of a wide variety of therapeutic misadventures. Several cases, many of them fatal, have been described in which salt was used as an emetic as “treatment” of accidental or deliberate drug overdose.22–25 Salt should never be used for this purpose. As pointed out by DeGenaro: “Salt may be an occasionally successful emetic. It is a reliable poison”.26 Other cases have occurred following the use of hypertonic saline, supposedly into the amniotic sac but with presumed systemic absorption, to induce an abortion.27 Fatal hypernatraemia occurred in a 7 year old boy in whom hypertonic saline was used as a scolicidal agent to irrigate hepatic hydatid cysts.28 Cholestyramine, an anion exchange resin, occasionally causes hypernatraemia and hyperchloraemia if the treated infant develops diarrhoea and vomiting, particularly if the diet is relatively high in solute.29
Salt poisoning has also been described when an excess of salt is accidentally administered, usually being mistaken for sugar in the preparation of feeds or oral electrolyte solutions. The most famous case occurred in Binghampton General Hospital, New York State, in 1962.30 In this disaster, several batches of infant formula were made up with salt instead of sugar; 14 infants received the hypertonic feed, six of whom died. The highest recorded plasma sodium concentration was 274 mmol/l, surprisingly in an infant who survived. Autopsy studies of the dead infants showed diffuse haemorrhagic encephalopathy in all cases. Similar sporadic cases have been described occurring both at home and in hospital. In one interesting case report, twin boys aged 14 months developed severe hypernatraemia (plasma sodium concentrations of 182 and 179 mmol/l, respectively), apparently having ingested salt voluntarily after being given a carton of table salt to play with in their cot.31 Reviewing this report after 41 years, however, raises questions about how the boys came to ingest the salt, there having been no witnesses to the event. I know of no other reported case in which infants or children have become hypernatraemic as the result of voluntary, unaided ingestion of salt; however, there is a single case report of an 85 year old woman, described as “mildly demented”, who became hypernatraemic after consuming the contents of one or two salt shakers, and having a history of consuming salt in the past.32
Deliberate or non-accidental salt poisoning was described in a series of 12 infants by Meadow in 1993.33 Several sporadic cases had been described earlier, and a further two were included in a series of 41 children suffering from illness induction syndrome from Great Ormond Street Hospital, London.34 The cardinal clinical features as described by Meadow are hypernatraemia (often recurrent) with high urinary sodium concentration, absence of evidence of renal, endocrinological or other organic disease that might explain the hypernatraemia, correction of hypernatraemia with separation from the perpetrator, and circumstantial evidence supporting the suspicion of deliberate poisoning with salt. Other forms of fabricated or induced illness were present in some but not all of the infants. In seven of Meadow’s original cases, and most of the other published instances, the perpetrator (usually the mother) admitted the abuse and explained how it was done. The route of administration is usually oral, either by adding salt to the feed or putting salt directly into the child’s mouth. Cases have also been described in which salt was administered via a nasogastric tube or even by interference with an intravenous infusion in hospital.
Management of salt overload
The evidence base for any particular approach to treatment is weak and entirely anecdotal. In contrast to hypertonic dehydration, the circulation is not usually contracted and there is no reason to give isotonic fluid. Logic suggests that the plasma sodium concentration should be lowered gently, as for hypertonic dehydration, and for the same reason. There are two reports of peritoneal dialysis being used to remove excess sodium,35,36 but the reasons for this are not clear from the published cases and conservative management might have been equally effective. The use of diuretics would seem logical, if the sodium concentration is slow to fall with appropriate fluid therapy alone; however, since very few paediatricians have treated more than one or two cases, controlled studies of different protocols are entirely lacking.
Differentiation between salt poisoning and other causes of hypernatraemia
Hypernatraemia caused by renal water loss due to DI and related disorders (see above) should be suspected from the typical history and evidence of impaired urinary concentrating ability. It is sometimes suggested that mineralocorticoid excess may be a cause of hypernatraemia, but this is probably incorrect. In a large study of patients with primary hyperaldosteronism, Brown and colleagues found that most of 489 measured plasma sodium values fell within the normal range, although the mean was slightly higher than that for normal subjects (143.3 mmol/l in females, 143.6 mmol/l in males) with none being above 147 mmol/l.37 This accords exactly with the values described in an earlier review by Relman.38 The cardinal electrolyte abnormalities in mineralocorticoid excess are hypokalaemia and metabolic alkalosis, not typical of salt poisoning.
The distinction between accidental and non-accidental salt poisoning cannot be made on clinical or physiological grounds, since the end result of both is the same. Only a meticulous evaluation of the history and the attendant circumstances of the case can resolve this, and doctors are not necessarily the professionals in the best position to obtain this information. The two conditions that should be distinguishable on clinical and physiological grounds are hypernatraemic dehydration and salt overload (however induced).
History and clinical findings
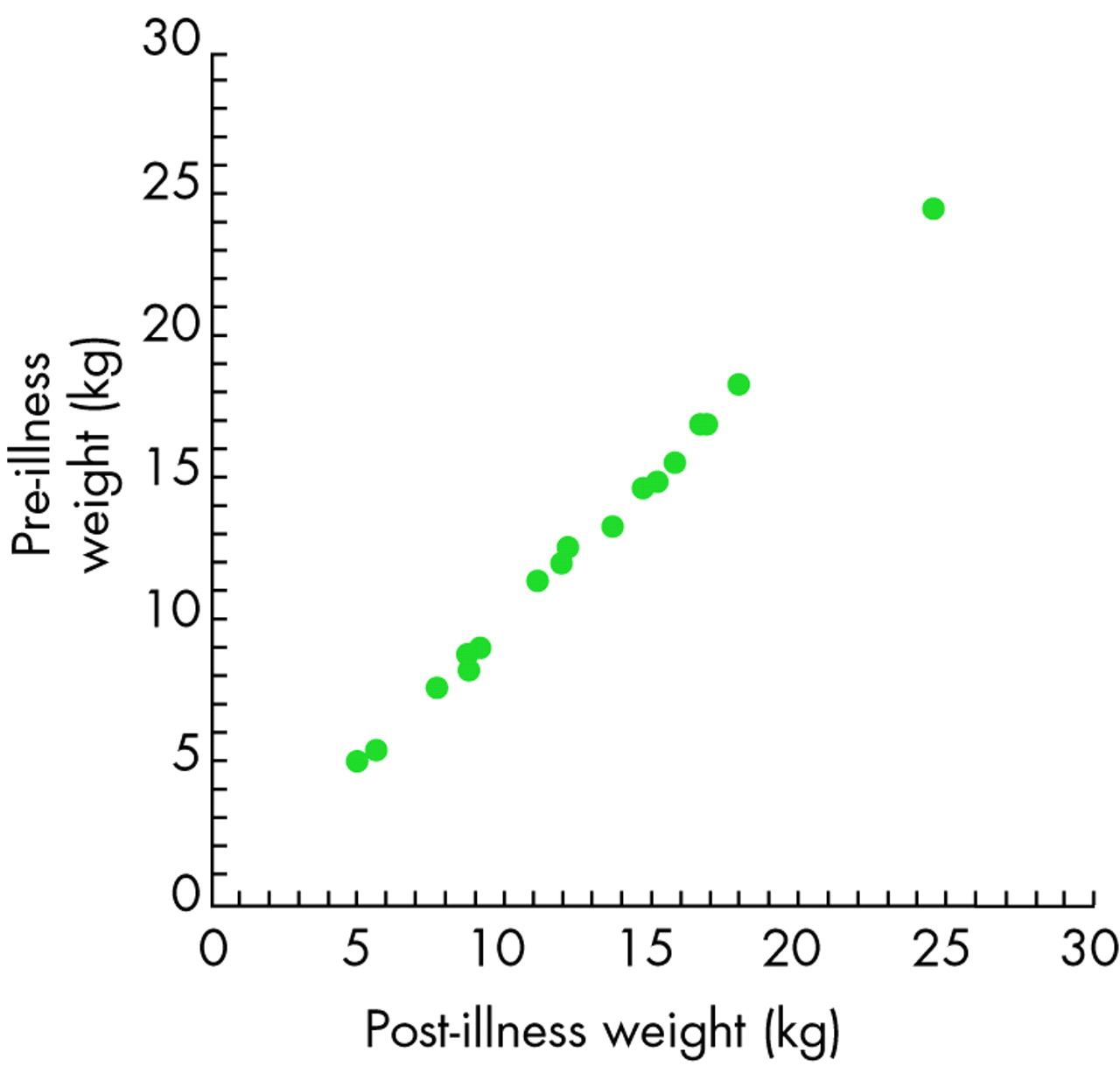
Changes in intracellular and extracellular fluid volume and tonicity in different types of hypernatraemia are shown in fig 1.39 By definition, patients with hypernatraemic dehydration are water depleted, usually severely so. As mentioned earlier, the magnitude of water loss required to produce a given rise in the plasma sodium concentration should be calculated on the basis of total body water, not ECF water. For example, an infant weighing 6 kg has a total body water of about 3.9 litres. To raise the plasma sodium concentration from 140 to 180 mmol/l from dehydration alone would require a water loss of about 860 ml, or 14% body weight. Moreover, total body sodium is actually reduced in hypertonic diarrhoeal dehydration since the primary cause is intestinal loss of hypotonic fluid, not pure water, so that the actual water loss in real cases would be somewhat higher than this estimate. It follows that dehydrated infants will have sustained a large weight loss compared with their pre-morbid weight. Unfortunately, although (one hopes) any sick infant will be weighed on admission to hospital, there is not always a recent recorded weight for comparison. However, it has been shown in careful reconstitution balance studies that, in dehydrated infants and children where the pre-illness weight was documented, there is excellent agreement between this and the weight achieved after appropriate rehydration therapy (fig 2),40 and this can reliably be used to give a retrospective estimate of the degree of weight loss at presentation. Infants who have been poisoned with salt will not show a weight loss unless there has been copious vomiting following salt administration. Again, a careful history is indispensable because the main cause of hypernatraemic dehydration in infancy is diarrhoea, not vomiting.

Changes in volume and tonicity of the major body compartments in normal conditions and three types of hypernatraemia. Open circles represent extracellular fluid (ECF) solute (mostly sodium chloride) and closed circles represent intracellular solute. Note that tonicity, shown as the degree to which solute particles are “packed” together, is always the same in the two compartments because water moves freely to osmotic equilibrium. The shaded area in the upper part of the extracellular compartment is the plasma component of the ECF. Panel A shows normal conditions. Panel B shows the effect of pure water loss (for example, diabetes insipidus or heat exhaustion). Panel C shows the effect of hypotonic sodium loss (for example, hypertonic dehydration). Panel D shows the effect of hypertonic sodium gain (for example, infusion of hypertonic sodium salts or salt poisoning). Adapted from Adrogué and Madias39 with permission.

{kind=link}
{kind=link}
Correlation between pre-illness and post-illness weight in 19 children with diarrhoeal dehydration in whom complete weight information was available. The correlation coefficient is 0.9988. Reproduced from Gorelick et al40 with permission.
Renal function
Another consequence of dehydration is reduced renal perfusion with a rise in the urea and creatinine concentrations (prerenal azotaemia). In Meadow’s series of salt poisoned infants, and in other reported cases where the relevant measurements were obtained, these were usually normal or low, indicating ECF volume repletion and well maintained renal perfusion.
Acidosis
Metabolic acidosis is a consistent feature of hypernatraemic dehydration, and it has been claimed that this finding favours the diagnosis of dehydration over salt poisoning. However, information on acid-base status was included in nine reports of accidental salt poisoning in the literature: the mean bicarbonate concentration was 11.1 mmol/l, with a range of almost zero (base deficit 26 mmol/l) to 18 mmol/l. Furthermore, hypernatraemia induced in rabbits by infusion of hypertonic saline caused severe metabolic acidosis.41 Finberg also pointed out that severe salt poisoning usually causes a profound metabolic acidosis, presumably due to release of hydrogen ions from desiccated cells.42 Acidosis does not therefore discriminate between the two conditions.
Urinary sodium concentration
All Meadow’s cases of salt poisoning, and those reported by others when it was measured, had high urine sodium concentrations, typically 200–230 mmol/l. This is entirely predictable, since the renal response to salt overload is to excrete the excess as rapidly as possible. However, the converse assumption, that infants with hypernatraemic dehydration have low urinary sodium concentrations, is not borne out by the (admittedly limited) published evidence. In 10 infants with hypernatraemic dehydration caused by gastroenteritis reported by Weil and Wallace,43 the mean urine sodium concentration was 98.5 mmol/l (range 35–232 mmol/l). Finberg induced hypernatraemia in dogs by dialysing them against hypertonic saline.44 In one group of nine dogs, constant weight (CW) was maintained by infusion of saline; in another group of eight dogs, dehydration (D) was allowed to occur (weight loss ⩾ 8% body weight). Comparable degrees of hypernatraemia were achieved in the two groups (CW 197 mmol/l, D 198 mmol/l). The mean urine sodium in CW dogs was 166 (108–215) mmol/l, and in D dogs 204 (26–280) mmol/l. The paucity of published information on urine composition in dehydrated infants is presumably due to the fact that oliguria makes urine collection very difficult in this group. Nevertheless, it is clear that urine sodium concentration alone does not reliably discriminate between dehydration and salt overload in the absence of dehydration. The reason for this is that the severe volume depletion typical of hypernatraemic dehydration causes maximal renal water conservation with the production of highly concentrated urine of small volume; the urine sodium concentration may be high even though the urine sodium excretion rate is low.
Fractional and actual sodium excretion
Direct measurement of sodium excretion rate is seldom practicable in these severely ill infants, especially if oliguria is present. Calculation of the fractional sodium excretion rate (FENa) is a good proxy, and this can in fact be converted to a reasonable estimate of sodium excretion rate if glomerular filtration rate is known. FENa is the sodium excretion rate divided by the sodium filtration rate, and reduces to the formula:

where the subscripts Na and Cr denote sodium and creatinine, respectively. The derivation of the formula is given in full in an annex to a recent related publication.45 Approximately simultaneous blood and urine samples are all that is required: the urine sample does not need to be a timed collection. To illustrate, an 18 month old child has a glomerular filtration rate (GFR) of about 50 l/day (36 ml/min). If the plasma sodium is 140 mmol/l, the daily filtered load of sodium is 50×140 mmol/day or 7283 mmol/day. The urinary sodium excretion rate, in health, is determined by the dietary intake which for a small child should not exceed 50 mmol/day (3 g of salt). Thus FENa in this situation is 50/7283 or 0.86%. If renal function and sodium intake are both normal, FENa is generally < 1%. It rises to high values in two situations: if GFR is reduced, either acutely or chronically, or if a sodium load is being excreted. The urine sodium:creatinine concentration ratio changes in the same direction as FENa but will be less discriminating if plasma creatinine is notably raised, as is often the case in hypernatraemic dehydration. It is hard to imagine a clinical situation in which the urine sodium and creatinine are known and the plasma sodium and creatinine are not.
Sodium excretion rate (ENa) can be calculated from FENa, GFR and PNa as follows:
ENa = FENa × GFR × PNa
Inspection of the terms from which the right hand side of this equation is derived will disclose that it reduces to ENa = UNa × V where V is urine flow rate, which is empirically correct. Thus, if the terms on the right of the equation are available (urine and plasma sodium and creatinine and body length or height) a reasonable estimate of sodium excretion rate can be derived.
How does body sodium relate to sodium excretion rate?
The kinetic model of Strauss and colleagues, based on studies in normal men, shows that sodium excretion rate is a function of the amount of sodium in the body above a “set point” at which the subject is in sodium balance on zero intake.46 If total body sodium is below this amount, sodium excretion virtually stops. Above this amount, there is a linear relation between “surplus” sodium and 24 hour sodium excretion rate. This relationship was revisited by Walser who re-analysed Strauss’s data and that from 12 other published studies.47 He confirmed the validity of the Strauss model and demonstrated a high degree of consistency among the studies considered. Following a step change from a lower to a higher sodium intake, the half time for excretion (the time required to excrete half the increased intake) is about 21 hours. It is predictable, therefore, that in a patient with hypernatraemic dehydration, in whom total body sodium is reduced, the sodium excretion rate will be minimal (whether expressed as FENa or calculated excretion rate as given above). In contrast, the individual with a salt overload will have a high sodium excretion rate. This is confirmed in actual measurements in a small number of infants with the two conditions. The available database is very small because the relevant urinary chemical analysis has very rarely been undertaken. The main points distinguishing salt overload from hypernatraemic dehydration are summarised in table 2.
Differences between hypertonic dehydration and salt overload. See text for further details