Article Text

Abstract
Background: The clinical response of patients with severe acute respiratory syndrome (SARS) to a combination of lopinavir/ritonavir and ribavirin was examined after establishing the in vitro antiviral susceptibility of the SARS associated coronavirus to a panel of antiviral agents.
Methods: The in vitro susceptibility of the prototype of SARS associated coronavirus to a panel of nucleoside analogues and protease inhibitors currently licensed for clinical use was studied. Forty one patients with SARS followed for 3 weeks were treated with a combination of lopinavir/ritonavir and ribavirin. The clinical progress and virological outcomes were monitored and compared with 111 patients treated with ribavirin only who served as historical controls.
Results: In vitro antiviral activity against SARS associated coronavirus was demonstrated for lopinavir and ribavirin at concentrations of 4 µg/ml and 50 µg/ml, respectively, only at 48 hours. The adverse clinical outcome (ARDS or death) was significantly lower in the treatment group than in the historical controls (2.4% v 28.8%, p<0.001) at day 21 after the onset of symptoms. The adverse outcome remained significantly lower in the treatment group than in the controls—both those diagnosed early (p<0.001) and those diagnosed later in the course of the epidemic (p = 0.002)—but there was no significant difference in adverse outcome rates between the two time periods (p = 0.548). No time related difference in outcome was observed in the control groups. A reduction in steroid usage and nosocomial infections was seen in patients initially treated with lopinavir/ritonavir, and these patients had a decreasing viral load and rising peripheral lymphocyte count. Multivariate analysis showed that age, hepatitis B carrier status, and lack of treatment with this antiviral combination were independent predictors of an adverse outcome. Lopinavir/ritonavir treatment was associated with a better outcome even when adjusted for baseline lactate dehydrogenase level.
Conclusions: The apparent favourable clinical response with lopinavir/ritonavir and ribavirin supports further randomised placebo controlled trials in patients with SARS.
- severe acute respiratory syndrome (SARS)
- coronavirus
- lopinavir/ritonavir
Statistics from Altmetric.com
Severe acute respiratory syndrome (SARS) is caused by a novel coronavirus.1–3 It has satisfied Koch’s postulations for causation by its consistent isolation from patients suffering from SARS, isolation of the virus and reproduction of disease in non-human primates after inoculation, and the presence of a specific antibody response against the virus in both SARS patients and artificially infected primates.4 In an earlier prospective study we suggested that there is an initial viral replicative phase followed by an immunopathological phase and end organ damage phase.5 We hypothesise that the immunopathological response is triggered by the viral antigen; the most strategic treatment is therefore to stop the viral replication at the beginning so that the peak viral load and the subsequent immunopathological damage will be minimised. An effective antiviral treatment is therefore required urgently. At present the treatment recommendations are largely empirical, ranging from supportive therapy without intervention to intensive immunomodulation with steroids. Controlled studies are difficult to perform in an epidemic of such a life threatening condition.6 We report the findings of an open trial of a combination of a protease inhibitor and a nucleoside analogue against a historical control group.
METHODS
Laboratory studies
The prototype virus (HKU-39849 isolate) was used for in vitro antiviral susceptibility testing. Initial screening was performed in 96-well microtitre plates seeded with fetal rhesus kidney-4. Doubling dilutions of antiviral agents starting at four times the peak serum concentration after a standard therapeutic dose down to 25% of the trough serum concentration were tested in quadruplicate against 100 TCID50 (median tissue culture infectious dose) of SARS coronavirus. A corresponding set tested with the drugs but without virus challenge were used as controls. The cells were scored for inhibition of the cytopathic effect at 48 hours. The antiviral agents tested included acyclovir, ganciclovir, cidofovir, foscarnet, ribavirin, interferon α, amantadine, zidovudine, stavudine, nevirapine, abacavir, ritonavir, and lopinavir.
Agents with detectable activity at the tested concentration were re-tested in a quantitative plaque reduction assay, followed by a chequerboard synergy test for various combinations of the antiviral drugs. Briefly, 24-well tissue culture plates with a confluent cell monolayer (1×105 cells per well) in 1.0 ml of minimal essential medium (MEM) with 10% fetal calf serum (FCS) were prepared. After the medium was aspirated, 50–100 plaque forming units (PFU) of SARS associated coronavirus (in 1% FCS/MEM containing the antiviral agents at appropriate concentrations) were added to each well. Plates were incubated for 2 hours at 37°C in 5% CO2. The inoculum was aspirated and 1.0 ml of overlay (1.0% low melting point agarose in 1% FCS/MEM with corresponding drug dilutions) was added to each well. Plates were further incubated for 48 hours at 37°C in 5% CO2. Cells were fixed by adding 2 ml 10% formaldehyde and the plates were incubated at room temperature for 2 hours. The agarose plugs were aspirated and each well was stained with 0.5% crystal violet prepared in 70% methanol. The viral plaques were counted. A chequerboard assay of inhibition of the cytopathic effect by the combination of ribavirin and lopinavir was performed using 25, 50, and 100 TCID50 per well in 96-well microtitre plates.
Clinical study
Between 24 March and 28 April 2003, 152 consecutive patients with probable SARS (according to the World Health Organisation definition) admitted to the United Christian Hospital and Caritas Medical Centre were recruited to the study after approval by the ethics committees. Once the diagnosis of SARS was established, ribavirin was given for 14 days (4 g oral loading dose followed by 1.2 g every 8 hours, or 8 mg/kg intravenously every 8 hours if the patient could not tolerate oral treatment) with a reducing regimen of corticosteroid for 21 days (starting dose: hydrocortisone 100–200 mg every 6–8 hours or methylprednisolone 3 mg/kg/day). Pulses of intravenous methylprednisolone (0.5–1 g/day up to 4 g) were used if patients developed increasing shortness of breath, oxygen desaturation, or radiological worsening.7
Patients treated before 16 April 2003 served as historical controls (n = 111). The clinical features and virological findings of the first 75 cases have been reported previously.5 After 16 April 2003, newly diagnosed SARS patients and patients who had not developed acute respiratory distress syndrome (ARDS) were started on a combination of lopinavir (400 mg)/ritonavir (100 mg) orally every 12 hours for 14 days (n = 41) after obtaining informed consent (ritonavir inhibits the CYP3A mediated metabolism of lopinavir and thereby potentiates the serum level of lopinavir).
Both the historical controls and the lopinavir/ritonavir treated group were given ribavirin and corticosteroid according to the same protocol. The primary outcome measure was a composite adverse outcome at 21 days, which was defined as severe hypoxaemia (meeting the ARDS criteria of arterial oxygen tension (Pao2) to fraction of inspired oxygen (Fio2) of <200 mm Hg8) or death. The treatment group was divided into two subgroups for further analyses. In the “initial treatment” subgroup (n = 12) lopinavir/ritonavir was started before pulse methylprednisolone (median time from onset of symptoms 3.5 days), while in the “rescue treatment” subgroup (n = 29) lopinavir/ritonavir was started after pulse methylprednisolone (median time from onset of symptoms 14 days). Clinical measurements and blood tests were performed daily.
Diagnosis of coronavirus infection was made from nasopharyngeal swabs on admission and convalescent serum on days 14–28 after onset of symptoms. In six randomly selected patients in the initial treatment subgroup and 12 historical controls with initially positive nasopharyngeal specimens for coronavirus tested by reverse transcriptase polymerase chain reaction (RT-PCR), quantitative RT-PCR was performed serially on days 5, 10, 15, and 20 after onset of symptoms. Stools were collected from all 41 patients in the treatment group for RT-PCR analysis at days 7, 14, and 21. The virological diagnostic protocol and other microbiological work up was performed in the same way as in our previous publications.1,5
Statistical analysis
All time related data were calculated from the day of symptom onset. The baseline characteristics of the historical controls and the treatment group were compared by Fisher’s exact test for categorical variables, Student’s t test, or Mann-Whitney U test for continuous variables where appropriate. The Kruskal-Wallis test was used to compare the cumulative doses of methylprednisolone in the various treatment subgroups. As this was an open non-randomised study, an imbalance in the baseline characteristics of the two groups was expected. To adjust for this imbalance, factors that were identified on univariate analysis to be associated with an adverse outcome at 21 days were analysed by multiple logistic regression with forward selection to identify independent predictive factors. In addition, the odds ratio for lopinavir/ritonavir treatment with respect to the primary outcome was adjusted for the baseline lactate dehydrogenase (LDH) level by multiple logistic regression, based on an a priori assumption that LDH might affect the outcome. As lopinavir/ritonavir was used later in the course of the epidemic, it was possible that any apparent benefit seen might be due to the experience gained by medical staff in treating the patients or attenuation of the virus during the epidemic. To determine whether there was a time related difference in outcome, the historical control group was divided into two subgroups according to admission date (an earlier and a later period) for further analyses. The date of division was arbitrarily chosen to achieve a reasonable balance of number of cases across the subgroups. The outcome of the two historical subgroups and the treatment group was compared using the χ2 test. A two tailed p value of <0.05 was considered significant. SPSS Version 11.0 (SPSS Inc, Chicago, IL, USA) was used for all analyses.
RESULTS
In vitro antiviral susceptibility testing showed that the cytopathic effect of the SARS coronavirus was inhibited by lopinavir at 4 μg/ml and ribavirin at 50 μg/ml after 48 hours of incubation (fig 1). The inhibitory effect had worn off at 96 hours. None of the other drugs tested had any inhibitory effect. The results were confirmed by plaque reduction assay. Using the chequerboard assay for synergy, inhibition of the cytopathic effect was achieved down to a concentration of lopinavir 1 μg/ml combined with ribavirin 6.25 μg/ml only when the viral inoculum was reduced to 50 TCID50 or below.
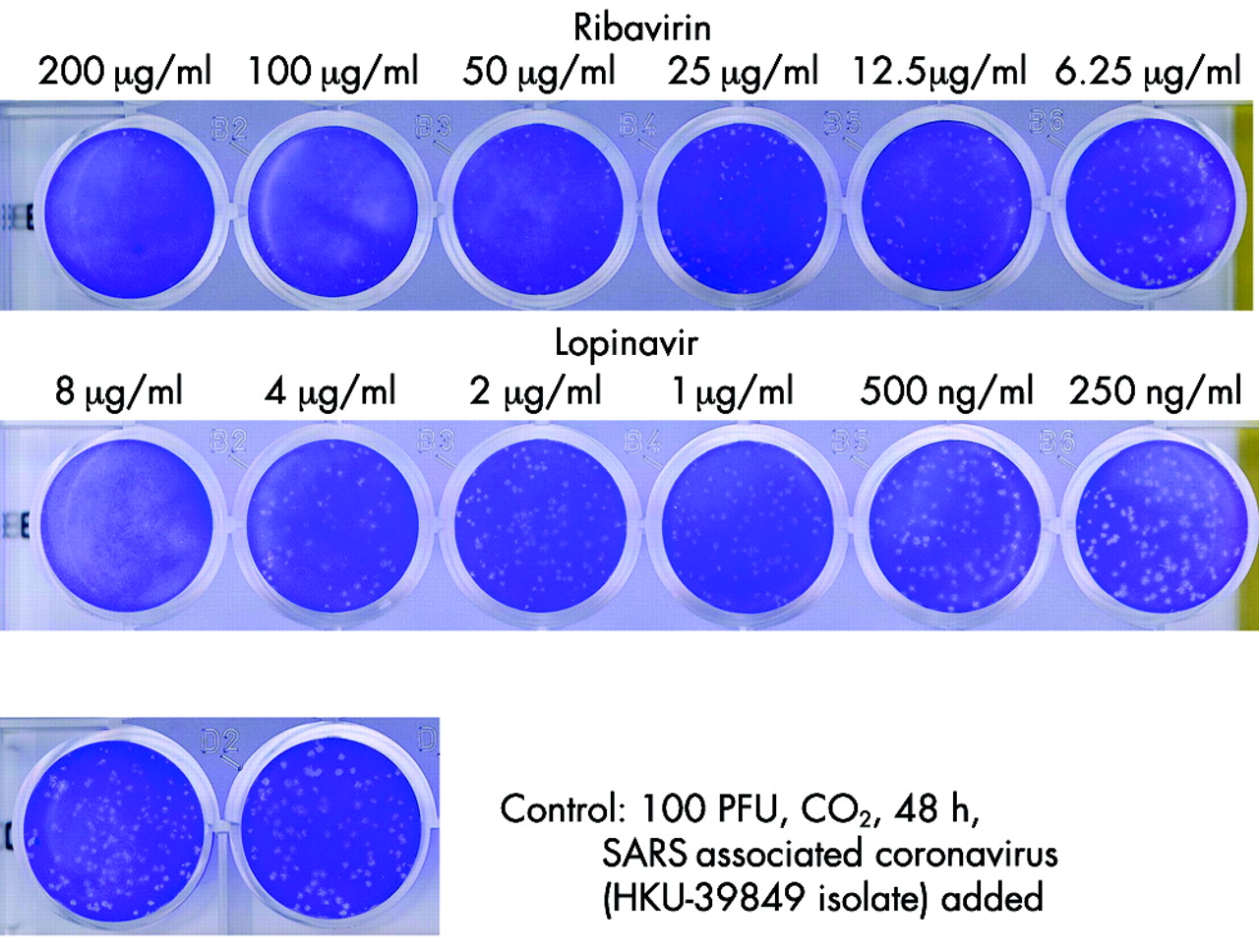
Dose dependent antiviral effects of ribavarin and lopinavir on SARS coronavirus. In vitro antiviral susceptibility testing showed that the cytopathic effect was inhibited by lopinavir at 4 μg/ml and ribavirin at 50 μg/ml after 48 hours of incubation.
The baseline characteristics and outcomes of the historical controls and the treatment group are shown in table 1. The 152 patients had a mean (SD) age of 41.4 (14.8) years and there were 58 (38.2%) men. 96.7% had virologically confirmed SARS associated coronavirus. Among the historical controls, 25 (22.5%) met the ARDS criteria of hypoxaemia and seven (6.3%) had died by day 21. In the treatment group only one patient (2.4%) met the ARDS criteria for hypoxaemia and there were no deaths. The 21 day adverse outcome rate was therefore 28.8% for the historical controls and 2.4% for the treatment group, giving an effect size of 26.4% (95% confidence interval 16.8 to 36.0, p<0.001) for lopinavir/ritonavir treatment.
Baseline characteristics and 21 day adverse outcome (death or development of ARDS requiring intensive care) of historical controls and treatment group
There was no significant difference between the two groups with respect to age and co-morbidity. However, the treatment group had fewer men, a higher initial platelet count, and lower initial LDH levels. Two models of multiple logistic regression were used to asses whether lopinavir/ritonavir treatment was independently associated with improved outcome. In the first model, multiple logistic regression with forward selection showed that age (p = 0.013), chronic hepatitis B infection (p = 0.007), and lack of lopinavir/ritonavir treatment (p = 0.011) were independently associated with the 21 day adverse outcome (table 2). In the second model, the odds ratio of lopinavir/ritonavir treatment was adjusted for the LDH level by multiple logistic regression, based on the a priori assumption that LDH might confound the outcome. Lopinavir/ritonavir treatment was found to be associated with a significantly better outcome (p = 0.014), despite adjustment for the LDH level (table 3).
Independent risk factors predicting adverse outcome of death or development of acute respiratory distress syndrome (ARDS) requiring intensive care within 21 days
Adjustment of odds ratio of lopinavir/ritonavir treatment for lactate dehydrogenase (LDH) level with respect to the adverse outcome of death or development of acute respiratory distress syndrome (ARDS) requiring intensive care within 21 days
To determine whether there was a time related difference in outcome, the control group was divided into two subgroups according to the date of admission. Period 1 was from 24 March to 27 March 2003 (4 days, n = 68) and period 2 was from 28 March to 15 April 2003 (19 days, n = 43). The dates were chosen to achieve a reasonable balance in the number of cases in each subgroup. Lopinavir/ritonavir was used for patients admitted between 16 April and 28 April (13 days, n = 41). The treatment group had a significantly better 21 day outcome than the two control subgroups with only one patient (2.4%) developing ARDS and no deaths (p<0.001 v period 1, p = 0.002 v period 2, χ2 test), but the 21 day outcome rate did not differ significantly between the two time periods in the historical control group (21 (30.9%) developed ARDS or died within 21 days in period 1 compared with 11 (25.6%) in period 2, p = 0.548, χ2 test).
The viral load in the nasopharyngeal swabs showed a progressive decrease in the initial treatment subgroup. A consistent decrease in the geometric mean viral load from 4.7 × 104 copies per ml on day 5 to an undetectable level on day 10 was seen in five out of six randomly selected patients (fig 2A). Case 6 had a transient decrease in viral load at day 10 followed by a rebound at day 15. This was associated with the use of pulse methylprednisolone on day 7. In contrast, 12 randomly selected patients from the historical control group showed an inverted V-shaped curve, with a geometric mean peak viral load of 1.9 × 107 copies per ml at day 10 which remained detectable (geometric mean 6.3 × 103 copies per ml) in 10 patients at day 20 (fig 2B). The stool RT-PCR positivity rate at day 21 was 2.4% for the treatment group and 67% for the historical controls.5

{kind=link}
{kind=link}
(A) Change in viral load by sequential quantitative RT-PCR for SARS associated coronavirus in nasopharyngeal swabs of six patients in the initial treatment subgroup. Note that case 6 was given pulse methylprednisolone on day 7. (B) Change in viral load by sequential quantitative RT-PCR for SARS associated coronavirus in nasopharyngeal swabs of 12 patients in the historical control group.
With regard to the progression of disease symptomatology in the treatment group compared with the historical controls, diarrhoea (24.4% v 62.2% p<0.001), recurrent fever (39% v 60.4% p = 0.0027), and worsening of the chest radiograph (51.2% v 81.1% p<0.001) occurred with reducing frequencies. The serial lymphocyte counts showed a rising trend in the initial treatment subgroup from 1.2 (0.7) × 109/l on admission to 1.0 (0.6) × 109/l, 1.3 (0.9) × 109/l, and 1.9 (1.1) × 109/l on days 7, 14 and 21 respectively, whereas in the historical control group the lymphocyte counts fell progressively from 1.1 (0.7) × 109/l on admission to 0.9 (0.1) × 109/l, 0.7 (0.1) × 109/l, and 0.5 (0.4) × 109/l on days 7, 14 and 21, respectively.
The cumulative pulse methylprednisolone dose did not differ between the historical controls and the treatment group (table 1). However, in the subgroup analyses the initial treatment subgroup required a significantly lower cumulative dose of pulse methylprednisolone than the rescue treatment subgroup and historical controls (median 0 g, 2.5 g, and 1.5 g respectively; p<0.001). There were also significantly fewer patients who suffered from nosocomial infections (0%) in the initial treatment subgroup than in the rescue treatment subgroup (27.6%) and historical control group (25.2%; p = 0.043 and p = 0.048, respectively). Patients with nosocomial infections received a higher mean (SD) cumulative dose of pulse methylprednisolone (2.6 (1.6) g) than those free of nosocomial infection (1.8 (1.5) g; p = 0.009).
Mild adverse reactions were experienced by 11 patients (26.8%) in the treatment group: gastrointestinal upset (n = 11), liver dysfunction (n = 7), headache (n = 6), blurred vision (n = 3). Only one patient required early discontinuation because of a significant rise in alanine aminotransferase (ALT) to more than twice the normal level. A fall in haemoglobin of more than 2 g/dl occurred in 29 patients (70.7%) in the treatment group, two of whom required a transfusion; 11 patients (26.8%) had asymptomatic bradycardia at a mean (SD) of 42 (6) beats per minute during the study period, which was similar to the historical controls.
DISCUSSION
In the early stages of the SARS epidemic before identification of the causal agent, histopathological changes in open lung biopsy specimens suggested the possibility of immunopathological damage.9 A broad spectrum antiviral agent (ribavirin) and immunosuppressive doses of steroids were therefore used as the empirical treatment. After the SARS associated coronavirus was isolated, in vitro antiviral susceptibility testing suggested that the virus could only be inhibited transiently by very high concentrations of ribavirin, levels difficult to achieve clinically. There was therefore an urgent need to find an alternative treatment. Our previous study on the sequential changes in viral load and disease progression suggested that there is an initial viral replicative phase which peaks at around day 10. This implies that there is a therapeutic window that could be exploited, provided an active antiviral agent was available. The key strategy in the treatment of SARS was therefore to find an effective antiviral agent that would decrease the peak viral load and thus the associated degree of immunopathological damage. This would decrease the need for immunosuppressants which were often associated with an increased risk of nosocomial infections, especially in patients who required mechanical ventilation.
Both the peak (9.6 µg/ml) and trough (5.5 µg/ml) serum concentrations of lopinavir can just reach the inhibitory concentration against the SARS virus.10 However, oral lopinavir/ritonavir may achieve a high fecal concentration because 20% of the drug is found unchanged in the stool.11 In our previous study we found that severe watery diarrhoea was a prominent feature in 73% of the SARS patients, and a high rate of fecal shedding was detected by RT-PCR between days 10 and 21.5 The gastrointestinal mucosa might therefore be an important reservoir for viral replication and dissemination. Although we may not achieve a satisfactory serum inhibitory concentration, the intestinal mucosa concentration may be high enough to stop viral replication at this site.
Because of the non-randomised nature of this open study, there were differences between the treatment and historical control group in terms of sex, platelet counts, and LDH levels. However, there was no significant difference between the two group with respect to age and co-morbidity, two important prognostic factors found in the previous study.5,12–15 On multivariate analysis, lopinavir/ritonavir treatment was an independent factor associated with improved 21 day outcome and this remained after adjustment for the LDH level. The 21 day adverse outcome rate of 2.4% for the treatment group compares favourably not only with our historical controls, but also with the reported figures of 21–23.3%12,13 and the projected figures of 13–43% for various age groups.16
Patients with SARS treated with lopinavir/ritonavir appeared to run a milder disease course in terms of diarrhoea, recurrence of fever, and worsening of chest radiographs. A reduction in the viral load was also seen (fig 2A). The viral load showed a progressive decrease in the initial treatment subgroup, in contrast to the expected inverted V-shaped curve previously reported.5 Moreover, the stool RT-PCR positivity rate at day 21 in the treatment group (2.4%) was markedly lower than that previously reported for the historical controls (67%).5 These findings suggest that the benefit of the combination was at least partly related to the antiviral activity.
In terms of adverse effects, there was little difference between the historical control group and that reported in the literature for lopinavir/ritonavir.11 It is important to note that the dose of ribavirin used in SARS is much lower than that used in the treatment of haemorrhagic fever. Thus, very few patients had their treatment terminated or suffered from major side effects except for anaemia. Furthermore, a fall in haemoglobin of 1.3 g/dl at a mean of 6.4 days was also found in a local cohort of patients with community acquired pneumonia (unpublished data).
As this is not a randomised study, there could be a few alternative interpretations of the results. Firstly, it is reasonable to suspect that the dramatic improvement in outcome in the treatment group could be due to the gain in experience in the management later in the epidemic. Attenuation of the virus during the course of an epidemic is another theoretical possibility to explain the improved outcome. Nevertheless, re-analysis of the historical controls, divided into subgroups according to an earlier and a later time of admission, did not change the conclusion that the treatment group had a better outcome than the controls in both periods. Moreover, there was no difference in the outcome between the two historical periods, suggesting that the period of admission was probably not the major reason for the apparent improved outcome in the lopinavir/ritonavir treatment group.
Secondly, corticosteroid use might confound the outcome as it might blunt the host response and promote viral replication. However, both the historical controls and the treated patients were put on the same protocol of corticosteroid treatment and the cumulative methylprednisolone dose did not differ between the two groups as a whole. The improved outcome in the treatment group was therefore not the result of reduced steroid use. In subgroup analysis, patients who received lopinavir/ritonavir as the initial treatment seemed to run a milder disease course and had a reduction in the viral load. Their need for rescue pulse methylprednisolone for severe respiratory deterioration was therefore reduced. Lopinavir/ritonavir might have improved the outcome either by a direct effect on the viral load or by an indirect steroid sparing effect because of a reduction in immunopathological damage.
Thirdly, the apparent improved outcome in the treatment group could be a result of a worse than expected outcome in the historical control group. Opponents of ribavirin and steroids have suggested that, in the USA where supportive treatment alone was used to treat SARS, the case fatality rate was zero.17 However, it is noteworthy that, in the USA, only eight out of 47 probable cases and none of the 162 suspected cases have had virological confirmation of SARS coronavirus infection.18 In contrast, in China, Taiwan, Canada, and Singapore there were 5327, 665, 251, and 238 cases of SARS, respectively, and case fatality rates of 7%, 27%, 17%, and 14% were reported.17
The use of high dose steroid treatment in SARS is, at best, controversial. Advocates for high dose steroid use have noted anecdotal successes of pulse methylprednisolone in SARS at doses used for the treatment of organ rejection.19 The same group of researchers investigated the response of patients with SARS to different steroid regimens and concluded that high dose pulse methylprednisolone therapy (organ rejection treatment range) appeared to be more efficacious and equally safe as a lower dose regimen.20 However, another group has not been able to show that the use of pulse steroid is associated with a better outcome.15 These findings need to be tested in randomised, placebo controlled studies which should include an arm not using steroids or antiviral agents.
In view of the apparent reductions in the composite adverse outcome at day 21, the viral load, the steroid dose, and the incidence of nosocomial infections, a randomised, placebo controlled study is justified to test the benefit of lopinavir/ritonavir in SARS. We propose that the combination of lopinavir/ritonavir and ribavirin should be tested against lopinavir/ritonavir alone and placebo.
Acknowledgments
The authors thank Dr E K Yeoh of the Hong Kong Government for facilitating the study and the staff of the Department of Microbiology, Queen Mary Hospital, the Department of Medicine and Intensive Care Unit, United Christian Hospital, and the Department of Medicine and Geriatrics, Caritas Medical Centre for their excellent technical assistance and patient management.
REFERENCES
Footnotes
-
↵* Members of the HKU/UCH SARS Study Group are listed at the end of the paper.
-
Conflict of interest: none declared.
-
Funding: Public Health Research Grant A195357 from the National Institute of Allergy and Infectious Diseases, USA, The Wellcome Trust Grant GR067072/D/02/Z, The University of Hong Kong and the Hospital Authority of Hong Kong, SAR.
-
Other members of the HKU/UCH SARS Study Group: B S F Tang, P C Y Woo, S K P Lau, Department of Microbiology, The University of Hong Kong; K I Law, Y H Hui, W S Leung, V L Chan, J S C Ng, United Christian Hospital; M W Tse, Caritas Medical Center.