Article Text

Abstract
Background and aims Evidence suggests that microbial communities in the preterm gut may influence the development of necrotising enterocolitis (NEC) and sepsis. Existing data often neglect fungi and whether bacteria were metabolically active or not. We sought to characterise the bacterial and fungal stool flora of preterm neonates and organism viability and evaluate any associations with NEC and sepsis.
Patients 136 stools from 32 patients (<32 weeks gestation) were collected between birth and day 95. Seven infants developed NEC and 13 sepsis.
Methods Stools were analysed by PCR-DGGE for assessment of the total bacterial and fungal communities by analysis of 16S rRNA and 28S rRNA, respectively. In 65 samples (25 infants), the viable (RNA) bacterial and fungal communities were analysed. Analyses were performed to examine the possible effects of demographic or treatment related factors and the development of NEC or sepsis.
Results 80 (66 viable) bacterial species were identified overall and 12 fungal (none viable). Total bacterial communities significantly differed between healthy infants and those with NEC or sepsis, with Sphingomonas spp. significantly associated with NEC. Significant drivers of community structure differed based on either total or viable analysis. Antifungal prophylaxis was associated with altered bacterial community and a reduction in bacterial richness was observed in week 4, correlating with high antibiotic exposure.
Conclusions Total and viable communities differ in preterm infants, and non-viable fungal species are present in infants on fungal prophylaxis. Exploration of viability and non-bacterial contributors to the total community may increase understanding of NEC and sepsis.
- Neonatology
- Microbiology
- Outcomes research
- Molecular Biology
Statistics from Altmetric.com
What is already known on this topic
-
Bacterial colonisation, particularly by Proteobacteria, appears to be a prerequisite for necrotising enterocolitis (NEC).
-
Invasive fungal infection is important in preterms and is caused primarily by Candida species.
-
The diversity of the gut microbiota in term neonates increases over time with community shifts attributable to diet changes and antibiotic treatments.
What this study adds
-
Even with almost universal antifungal prophylaxis, evidence of fungal colonisation exists but these species are not viable.
-
The viable bacterial community has a lower diversity but follows comparable shifts compared with the total bacterial community.
-
Sphingomonas spp. colonisation was significantly (p=0.0001) associated with NEC.
Introduction
Prematurity remains the leading predisposition to neonatal death and long term disability,1 with infection and necrotising enterocolitis (NEC) increasing in preterm infants.2 Associated mortality and long term consequences for survivors underpin the need for improved understanding and prevention of both prematurity and the associated morbidities.3 Recent interest has focused on the role of the gut microbiota in the pathogenesis of NEC and sepsis.4
The bacterial portion of the gut microbiota plays a crucial role in the development of immune function, micronutrient production and absorption, mucosal barrier function, and modulation of the systemic inflammatory response.5 Although bacterial colonisation is considered a prerequisite for NEC6 no single causative bacterial agent has been identified. Studies reporting specific bacterial associations with NEC suggest a role for Proteobacteria, commonly Enterobacteriaceae.7 ,8 Preterm neonates also show delayed colonisation by ‘healthy commensal’ organisms, especially bifidobacteria and lactobacilli, potentially attributable to medical management of prematurity.9 ,10 The diversity of the total gut microbiota in term neonates increases over time with abrupt community shifts associated with diet changes or antibiotic treatments.11 Total community analysis is based on DNA and will include both metabolically active (live) and dead microorganisms. Analyses based on the total gut microbiota provide a phylogenetic picture of the community, but do not reflect the viable community,12 which differs from the total community in adults.13 Viable community analysis is based on RNA and includes only metabolically active microorganisms.
The fungal community within the gut microbiota of preterm infants remains relatively unexplored, despite the importance of candidal infection in neonates14 and increasing antifungal prophylaxis within neonatal intensive care units (NICUs).15 ,16 How antifungal prophylaxis affects the neonatal gut microbiota remains largely unknown.
We aimed to examine total and viable bacterial and fungal communities in the gut of preterm infants exposed to current NICU practices, exploring potential associations of the gut microbiota with a diagnosis of sepsis or NEC, antifungal prophylaxis, gender, birth mode, gestational age, birth weight, and postnatal age.
Methods
Patients and samples
Preterm infants (<32 weeks gestation) cared for in the Royal Victoria Infirmary (Newcastle upon Tyne, UK) had stool collected for routine surveillance between March 2010 and January 2011. A total of 136 first and weekly stool samples from 32 infants between birth and day 95 were frozen at −20°C within 24 h of collection. In 65 samples (25 infants) a portion was preserved in RNAlater (Ambion, Texas, USA) and thus available for analysis of the viable community.
This unit has standardised feeding practices, antibiotic and antifungal use. Fluconazole is used as antifungal prophylaxis if <26 weeks/<1 kg and in NEC. Antibiotics are stopped at 48 h unless proven infection (see online supplementary data S1). Clinical information was obtained from notes: NEC was categorised independently by two clinicians (JEB/NDE) from notes, x-ray and operative findings and classified as surgical (diagnosis confirmed operatively) or medical NEC (un-equivocal pneumatosis). Sepsis was defined by positive blood culture, along with treatment for a minimum of 5 days and signs suggestive of infection.
PCR-DGGE analysis
Nucleic acid extraction was carried out on 100 mg of stool (wet weight). DNA was extracted from all samples for analysis of the total community using the PowerLyzer PowerSoil DNA Isolation Kit (MoBio, California, USA) according to the manufacturer's protocol. RNA was extracted from samples stored in RNAlater for analysis of the viable community using the PowerMicrobiome RNA Isolation Kit (MoBio) according to the manufacturer's protocol before further treatment with TURBO DNase (Ambion) to remove remaining contaminating DNA. The quality of the extracted DNA and RNA was determined by electrophoresis of a 5 μl aliquot on a 1% agarose gel and stained with SYBR Safe and SYBR Gold, respectively. Reverse transcription was carried out on all extracted RNA with Superscript II reverse transcriptase (Invitrogen, California, USA) using random hexamers (Qiagen, California, USA) and 40 U RNaseOUT (Invitrogen) according to the manufacturer's instructions. Validation of the methods was performed prior to commencing this study.
PCR amplification of the bacterial community targeted the V3 region within the 16S rRNA gene and was performed with primers V3F-GC and V3R as described by Muyzer et al,17 according to the methods of Baxter and Cummings.18 PCR amplification of the fungal community targeted the 28S rRNA gene and was performed with primers U1 and U2-GC,19 according to the method of Nelson et al.20
Denaturing gradient gel electrophoresis (DGGE) analysis was performed using the D-Code DGGE system (Bio-Rad, California, USA). For bacterial analysis, PCR products were loaded on to polyacrylamide gels (12%) with a denaturant gradient of 34%–55% denaturant (with 100% denaturant corresponding to 7 mol/l urea plus 40% v/v formamide). Gels were run at 60°C for 4.5 h at 200 V. Conditions for fungal DGGE were a denaturing gradient of 40%–60%, run at 60°C for 17 h at 70 V. Gels were stained for 30 min with SYBR Gold (Invitrogen) and viewed with UV transillumination using the Gel Doc 2000 gel documentation system (Bio-Rad). The analysis of the DGGE banding patterns employed TotalLab Phoretix 1D and 1D Pro software (Nonlinear Dynamics, Newcastle upon Tyne, UK). A ladder of amplicons derived from pure culture organisms was generated in house and loaded on each DGGE gel for multiple gel alignment (see online supplementary data S2).21 Due to the relative lack of diversity in the fungal DGGEs, the fungal ladder was used in the identification of fungal taxa by aligning bands from the sample lanes with known bands from the ladder lanes.
Specific bacterial DGGE bands were excised and eluted in 10 µl of molecular biology grade water and incubated at 4°C overnight. The full 10 µl of eluted DNA was amplified by PCR using the original V3F and V3R unclamped primer set,17 and excess dNTPs and primers were removed using ExoSAP-IT (USB, Ohio, USA). PCR products were then cloned using the pGEM-T easy vector (Promega, Wisconsin, USA) and plasmid DNA purified using the PureYield Plasmid Miniprep kit (Promega) per the manufacturer's protocol. Sequencing was carried out commercially using an ABI 3730XL sequencer (Eurofins MWG Operon, London, UK). BLASTn was used to determine the closest sequence homologies of each band (mean length 238 bp; range 166–457 bp) from the National Center for Biotechnology database.
Statistical analysis
Normalised DGGE data were subjected to canonical correspondence analysis (CCA) using CANOCO V.4.522 to analyse the association between the bacterial community and discrete (gender, diagnosis of sepsis and NEC, caesarean or vaginal birth, antifungal prophylaxis) and continuous (day of life, birth weight, gestation age) variables. Monte Carlo permutation testing under full model (499 permutations) was used to analyse the relationship between the discrete and continuous variables and the DGGE band matrix. A probability (p) value of <0.05 was deemed significant for all statistical tests. To describe the structure of the microbial communities present in the stool, Shannon diversity indices (H′) (accounting for both evenness and abundance) were calculated for each sample using the normalised DGGE band matrix. Species richness (based solely on the total number of taxa) was calculated based on the number of bands in the DGGE profile, where each band was assumed to be a single taxon. Statistical significance of bands associated with disease was determined using an unpaired t test.
Ethical approval
Initial collection was part of routine service. Ethical approval was obtained to include molecular techniques in August 2010. Prospective parental approval was obtained for all samples collected from December 2010.
Results
Clinical characteristics
Demographic information is provided in table 1 and details of all individual infant antibiotic exposure in online supplementary data S1. A total of 32 patients contributed to the total analysis: 25 patients contributing to viable analysis did not differ significantly demographically from the overall population. Overall, 30 infants (23 from viable subset) received some breast milk and 30 (23 from viable subset) received antifungal prophylaxis (fluconazole). None received probiotics or prebiotics. Seven developed NEC (three surgical); four contributed to the viability analysis. Six samples predated and 12 postdated NEC diagnosis. In all, 13 infants developed sepsis with five infants having more than one episode: 10 contributed to the viability analysis. In all, 22 samples predated and 32 postdated sepsis diagnosis. Organisms causing sepsis were: 10 coagulase negative staphylococci (CONS), one Staphylococcus aureus, two Enterococcus faecalis, two Escherichia coli, one Klebsiella pneumoniae, one Pseudomonas aeruginosa, one Micrococcus luteus and one Candida parapsilosis.
Demographic data and species richness from whole patient cohort
Total communities
DGGE identified 80 individual species (mean 14 per stool, range 2–26). Interestingly, mean numbers of total bacterial species did not differ among healthy, NEC and sepsis patients (table 1). The most prevalent bacteria belonged to the genera Enterococcus, Streptococcus and Escherichia. DGGE analyses identified 12 fungal species (mean 2 per stool, range 0–6). Half of infants in the cohort carried at least one fungal species, half showed no fungal colonisation. For most individual infants where fungi were identified they were in every sample with high intrapatient concordance. No fungal species were observed in any stool from any infant who developed NEC (all on fluconazole) (table 1). Candida spp. were the most abundant fungi (61%) with Candida albicans and Candida glabrata responsible for 30% and 29%, respectively.
Constrained ordination analyses demonstrate variation between samples attributable to external environmental variables. Grouping shows similarity between samples and the association of variables. Constrained ordination using CCA was carried out on the total bacterial community that explained 35% of the total variance (figure 1). The principal axis explained 19% of the variance separating patients according to disease state with healthy infants clustered separately from infants with NEC or sepsis. Patients with NEC (p=0.002) or sepsis (p=0.002) had significantly different profiles compared with healthy infants. Only colonisation with Sphingomonas spp. was significantly (p=0.0001) associated with NEC. Delivery mode (p=0.01) and gender (p=0.012) also influenced the bacterial community. Only two patients did not receive antifungal prophylaxis resulting in significantly different (p=0.03) bacterial profiles. Interestingly, the bacterial community was not significantly influenced by gestational age and birth weight.

CCA based on the total bacterial community. Comparison of patient profiles (Healthy patients (○), patients diagnosed with NEC (•), and patients diagnosed with sepsis (▪)) with discrete (▴) – N = NEC (P=0.002), S = sepsis (P=0.002), AF = antifungal treatment (P=0.03), CS = caesarean / Vag = vaginal birth (P=0.01), M = male / F = female (P=0.012) and continuous explanatory variables (- -▸) – GA = Gestation age (P=0.148), DOL = day of life (P=0.002). Sequenced bands (▵) include – Eb = Enterobacter, Es = Escherichia, Sp = Sphingomonas, Sa = Staphylococcus, Sr = Streptococcus.
The species richness in the total bacterial community increased with increasing postnatal age, but the richness of the fungal community remained relatively constant (see online supplementary data S3).
Viable communities
DGGE analyses on the viable bacterial community demonstrated the presence of 66 individual species (mean 6 per stool, range 1–14). There was no statistical difference between the numbers of viable bacterial species detected in infants with NEC or sepsis compared with healthy infants (table 1). The most abundant viable bacteria matched the total community (Enterococcus, Streptococcus and Escherichia spp.), but the constrained ordination differed (figure 2). While both CCAs explained the same variance in the first two axes (35%), only gestational age (p=0.002), day of life (p=0.004) and sepsis (p=0.004) had a significant effect on the viable community. Colonisation with Sphingomonas spp. remained significantly associated with NEC (p=0.0001). To identify any impact of differences in cohort size between total and viable communities, an additional CCA using DNA matched to the viable cohort was also performed. This confirmed the bacterial analysis on the full cohort, showing gender (p=0.002) and birth mode (p=0.012) to be significant and gestation age to be insignificant in shaping the gut microbiota (data not shown).

CCA based on the viable bacterial community. Comparison of patient profiles (Healthy patients (○), patients diagnosed with NEC (•), and patients diagnosed with sepsis (▪)) with discrete (▴) – N = NEC (P=0.188), S = sepsis (P=0.004), AF = antifungal treatment (P=0.144), CS = caesarean / Vag = vaginal birth (P=0.366), M = male / F = female (P=0.166) and continuous explanatory variables (- -▸) – GA = gestation age (P=0.002), DOL = day of life (P=0.004). Sequenced bands (▵) include – Bi = Bifidobacteria, Eb = Enterobacter, Es = Escherichia, Sp = Sphingomonas, Sr = Streptococcus.
No viable fungi were detected in any sample (table 1). To ensure this was not a methodological issue, cultured isolates of C albicans were spiked into stool and the RNA methodology followed. The C albicans species were successfully detected by PCR-DGGE.
Effects of increasing age on the total and viable bacterial communities
Total bacterial samples were matched with the corresponding viable sample to compare changes with increasing age. Diversity increased from week 1 of life, although this increase was not continuous, with fluctuations in the bacterial community structure occurring throughout the first 9 weeks (figure 3). Trends were similar for total and viable communities but the numbers of bacteria deemed viable were lower than that of the total community. Overall the diversity and richness of the samples increased over the first 9 weeks correlating with reduced antibiotic exposure as well as increasing age, but a reduction was noted in week 4 when the diversity was more established and antibiotic administration was still relatively high. The numbers of samples available each week were variable preventing further statistical analysis of this current cohort.

{kind=link}
{kind=link}
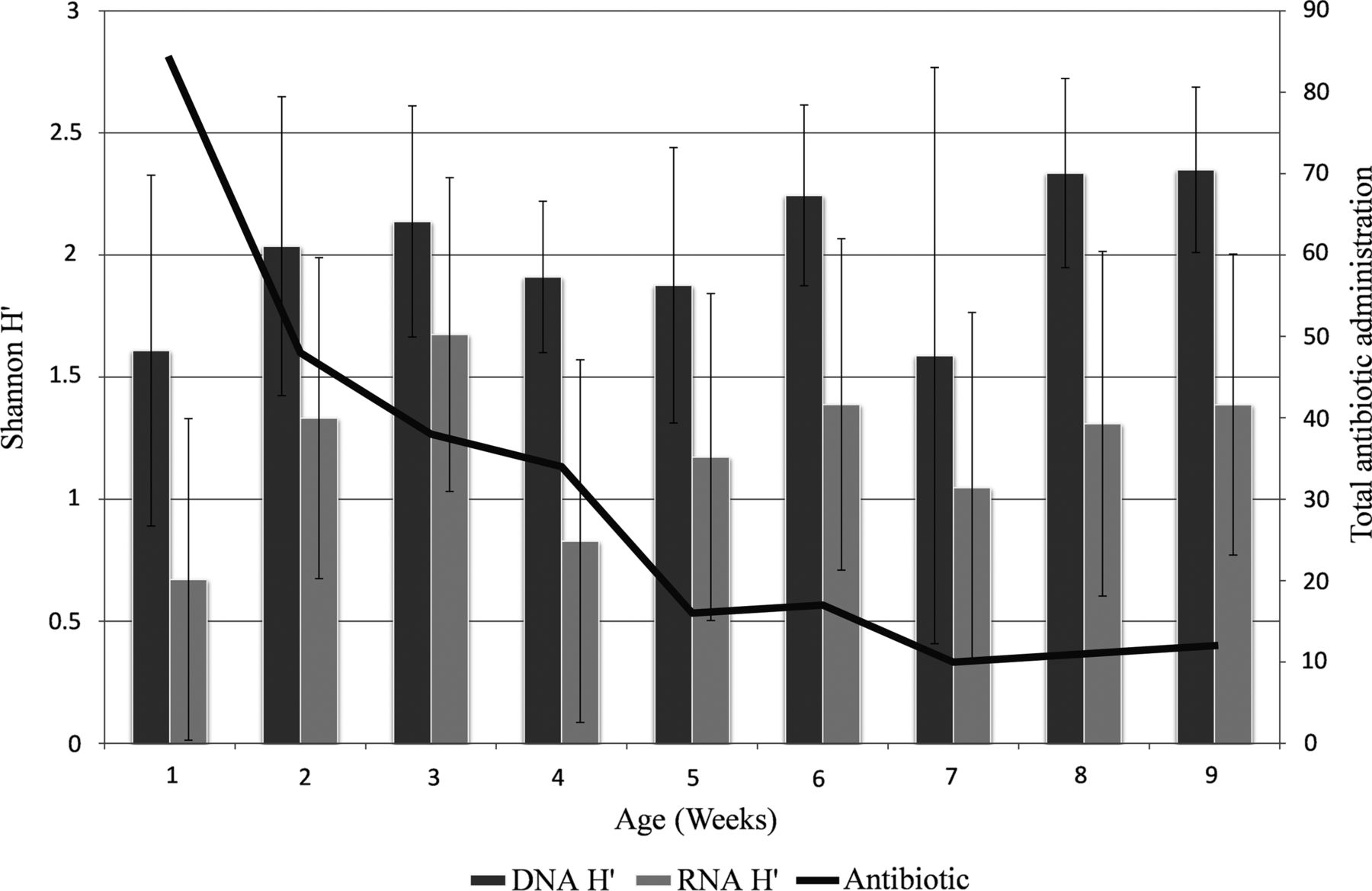{kind=link}
A comparison of the total and viable bacterial Shannon diversity (H') with the total number of antibiotics administered each week. Error bars represent the standard error of mean.
Discussion
We have explored two novel aspects of the preterm faecal microbiota: fungal colonisation and the role of organism viability. Total bacterial profiles of infants with NEC and total and viable profiles of infants with sepsis were significantly different from healthy infants, supporting a role for bacterial colonisation in the pathophysiology of these diseases.23 This is a descriptive study not a case-control study, and aimed to describe variables related to the development of the gut microbiota in preterm neonates over a period of time. Subsequent analyses are then subject to influence by practices within neonatal intensive care that confound associations, making the direction of effect unclear and the apparent effect of the disease states may not be causal. However, when examining samples predisease and postdisease diagnosis using unconstrained analysis we observe that samples before disease onset still group separately from healthy patients (see online supplementary data S4).24 This suggests differences in the gut microbiota predate the onset of disease.
Importantly and to our knowledge uniquely, Sphingomonas spp. colonisation was significantly associated with NEC in both the total and viable analysis. This genera belongs to the Proteobacteria phylum, recently reported to increase before NEC onset.6 ,7 Sphingomanads have previously been identified within biofilms formed in water supply systems25 although this would appear to be an unlikely route of colonisation within NICU where most exposure to water is to sterile water. Further work is needed to determine if the association of Sphingomonas spp. with NEC remains significant in a larger cohort and elucidates mechanisms of pathogenicity, and will include sampling the neonatal intensive care environment.
Unlike others24 ,26 we have shown no significant difference between the number of total bacterial species, whether total or viable, in populations of infants with NEC or sepsis compared with healthy infants. Differences in findings between studies may depend on the timing of sampling in relation to disease onset. Surprisingly gestation only influenced viable data, and birth weight was never found to have a significant effect on the gut microbiota, despite the well-recognised importance of these factors for the development of NEC.27–30 The most prevalent genus of bacteria identified in our total and viable analyses (Enterococcus, Streptococcus and Escherichia) agreed with those identified by others.1 ,9 ,31 ,32 Interestingly, Staphylococcus spp. were not found among the most prevalent species despite this genus contributing to the majority of positive blood cultures and being previously reported as a prevalent genus.10 ,33 This may relate to specific antibiotic preferences used within individual units.
Fungal colonisation was identified in half the infants but no viable fungi were detected in this cohort with almost universal fluconazole use. The stability of fungal profiles within individuals suggests that fungal DNA persists in the gut long after fluconazole prophylaxis due to its fungistatic properties successfully inhibiting replication. Although small in number (n=2), the infants who did not receive fluconazole had significantly different total bacterial profiles, although again this could be confounded by their clinical differences that resulted in them not requiring fluconazole. Fluconazole has direct antibacterial properties especially against Gram positives;34 there may also be community effects from liberating niches that would otherwise be occupied by fungi. Despite fluconazole use, identified fungal species were dominated by Candida spp.35 ,36 although these were non-viable. We also did not find a correlation between fungal colonisation and mortality or NEC.36
A major benefit of assessing the viable community rather than just the total community is that it potentially gives insight into taxa that are driving major metabolic activities and microorganism–host interactions, and thus may give insight into strategies to alter outcomes.12 ,13 However, the importance of separately assessing the viable community within preterm infants is currently unclear. We found important differences in the variables deemed to be significantly driving bacterial community structure based on either total or viable analysis. While sepsis was found to be significant in both analyses, gestational age was only significant for the viable community and gender and birth mode were only significant for the total community. Analyses incorporating the viable portion of the gut microbiota may gain increasing importance when assessing potentially important gut microbiota manipulations. Of current interest is the use of probiotics37 and lactoferrin;38 however, the mechanism of action and effect of such treatments on the gut microbiota require further research.39
LaTuga et al1 recently showed a low to moderate total bacterial diversity with a mean H′ of 1.02 from a cohort that was younger with a lower gestation compared with ours. We report relatively high total bacterial diversity (mean H′ of 2.04) but our associated viable bacterial diversity was lower with a mean H′ of 1.18. It has also been shown that antibiotic administration decreases the numbers of anaerobic bacteria in the gut microbiota, with counts of bifidobacteria particularly reduced.40 Our results support this with bifidobacteria being detected in 8.8% of the total community and only 3.1% of the viable community. From week 5 onwards both the total and viable community generally show an increasing diversity and richness, by which stage the majority of patients were no longer receiving antibiotic treatment.
Molecular based research into the association of the total bacterial community with NEC has increased our understanding of the disease: the complex multifactorial pathophysiology appears to be influenced by a variety of bacterial genera, individually or promoting shifts in communities. Faecal samples allow for non-invasive elucidation of the gut microbiota; however, the faecal microbiota may not precisely represent the gut microbiota.41 Employing molecular techniques allow greater coverage of the microbial community with only 20% of the gut microbiota reported to be cultivable.42 This may allow detection of important species not readily cultivated such as Spingomonas spp. here associated with NEC. High throughput next generation sequencing platforms, such as 454 pyrosequencing, are becoming increasing employed in clinical research due to the detection of low abundance taxa.43 However, as in DGGE, pyrosequencing is also subject to PCR bias.44
This novel study employing a relatively large cohort helps to further elucidate total as well as viable organisms of the gut microbiota in association with NEC and sepsis. We show abnormal bacterial colonisation in association with the development of NEC and sepsis, with colonisation by Sphingomonas spp. significantly associated with NEC. While antifungal prophylaxis significantly affects the total bacterial community, the presence of fungal species in the gut was not demonstrated to affect bacterial richness. Further work is needed to investigate the role of community microbial dynamics in the pathophysiology of NEC and infection, while additional exploration of the total and viable communities may add further to our understanding.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors CJS, NDE, JEB, SPC and JDP contributed to the study idea and design. JEB, ECLM, CJS and NDE contributed to collection of samples and data, CJS, AN, DS, CL and SPC contributed to analysis of samples and data. All authors have contributed to and reviewed the manuscript.
-
Funding This research was funded by the charity Tiny Lives who have played no part in the study design, in the collection, analysis and interpretation of data; in the writing of the manuscript; and in the decision to submit the manuscript for publication.
-
Competing interests None.
-
Ethics approval County Durham and Tees Valley.
-
Provenance and peer review Not commissioned; externally peer reviewed.