Article Text

Abstract
Background Neonatal sepsis due to intestinal bacterial translocation is a major cause of morbidity and mortality. Understanding microbial colonisation of the gut in prematurity may predict risk of sepsis to guide future strategies to manipulate the microbiome.
Methods Prospective longitudinal study of premature infants. Stool samples were obtained weekly. DNA was extracted and the V6 hypervariable region of 16S rRNA was amplified followed by high throughput pyrosequencing, comparing subjects with and without sepsis.
Results Six neonates were 24–27 weeks gestation at birth and had 18 samples analysed. Two subjects had no sepsis during the study period, two developed late-onset culture-positive sepsis and two had culture-negative systemic inflammation. 324 350 sequences were obtained. The meconium was not sterile and had predominance of Lactobacillus, Staphylococcus and Enterobacteriales. Overall, infants who developed sepsis began life with low microbial diversity, and acquired a predominance of Staphylococcus, while healthy infants had more diversity and predominance of Clostridium, Klebsiella and Veillonella.
Conclusions In very low birth weight infants, the authors found that meconium is not sterile and is less diverse from birth in infants who will develop late-onset sepsis. Empiric, prolonged antibiotics profoundly decrease microbial diversity and promote a microbiota that is associated not only with neonatal sepsis, but the predominant pathogen previously identified in the microbiome. Our data suggest that there may be a ‘healthy microbiome’ present in extremely premature neonates that may ameliorate risk of sepsis. More research is needed to determine whether altered antibiotics, probiotics or other novel therapies can re-establish a healthy microbiome in neonates.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Introduction
The pattern of enteric colonisation is thought to differ in premature infants in an intensive care setting when compared with colonisation patterns in healthy, term, breastfed infants.1,–,3 Culture-based studies of premature infants have identified delay in colonisation of bacteria, and predominance of potential pathogens including Enterobacteria, Bacteroides, as well as known pathogens, including Clostridia, Staphylococci, Pseudomonas and Klebsiella.1 Life-threatening complications of prematurity, including sepsis and systemic inflammatory response syndrome (SIRS) are related to microbial–host interactions, including direct bacterial translocation resulting in sepsis.4,–,7 Microbial colonisation patterns in immune naive premature infants may lay the groundwork for life-long disease risk from immune modulation.8,–,14 Maternal infections at the time of delivery, and at times, significant illness severity with premature birth, frequently prompt the use of early, broad-spectrum antibiotics in premature infants.5 ,15,–,17 Antibiotic use and delayed initiation of enteral feeding may influence the type and amount of micro-organisms colonising the intestinal tract.1 ,5 ,18 ,19 Despite guidelines of many units to decrease antimicrobial exposure and risk of neonatal infection,20 ,21 many premature infants are exposed to broad-spectrum antibiotics for prophylactic or prolonged empiric therapy; clinical complications associated with this exposure are beginning to be elucidated.5 ,16 ,22 ,23 In our most premature patients at high risk for morbidity and mortality (VLBW, very low birth weight infants, <1500 g), the availability of deep sequencing technologies have begun to inform a more complete understanding of a rich and diverse microbial landscape that may have profound implications for health.19 ,24,–,26
What is already known on this topic
Gut microbial acquisition in extremely premature infants in intensive care settings is altered by delayed feeding, antibiotic administration and hospital-acquired pathogens.
Neonatal sepsis can result from bacterial translocation from the intestines and is a significant cause of morbidity and mortality.
Comprehensive longitudinal evaluation of microbial colonisation with cutting-edge sequencing technology has not been applied to elucidate meaningful patterns that might highlight logical life-saving interventions.
What this study adds
Meconium in high risk, extremely premature infants is not sterile.
Broad-spectrum antibiotics profoundly affect gut colonisation with Staphylococcus predominance, identified comprehensively through deep sequencing, increasing risk of life-threatening sepsis with dominant pathogens.
Patterns emerge for healthy premature infants and infants who develop sepsis, providing data for potential time points for life-saving interventions to alter microbial colonisation.
The short- and long-term implications of perturbations of the developing microbiome in premature infants are largely unknown, and potential opportunities exist to alter and normalise the microbiome to ameliorate disease risk with, for instance, pre and probiotic therapies.27,–,30 The purpose of this study was to investigate the developing intestinal bacterial microbiome in VLBW infants at risk for sepsis, using culture-independent 454 pyrosequencing (high throughput sequencing targeted at the bacterial 16S rRNA gene). Rather than looking at one point in time, we observed VLBW infants over time with a focus on the colonisation patterns prior to late-onset sepsis in order to correlate colonisation patterns and changes in diversity with clinical factors.
Materials and Methods
Institutional Review Board approval for the study was obtained in April 2010 (CPHS 21761) and renewed in April 2011 from the Centre for Protection of Human Subjects at Dartmouth College and Dartmouth Hitchcock Medical Center, and parents provided written, informed consent for their infants' participation in the study. Approval was obtained in April 2010 (CPHS 21761) from the Centre for Protection of Human Subjects at Dartmouth College and parents provided written informed consent.
Inclusion criteria and clinical data
VLBW infants were enrolled within 2 days of birth for the study. Inclusion criteria included birth weight <1500 g without major anomalies. Clinical variables included maternal variables (including premature preterm prolonged rupture of membranes (PPPROM), maternal infection, gestational hypertension), feeding type (breast milk, fortified breast milk, or formula), antibiotic exposure and clinical complications including sepsis and SIRS (figure 1A). SIRS was defined as ≥2 of the following: (1) temperature >38.5°C or <36°C, (2) tachycardia (mean heart rate >2 SD above normal, (3) tachypnoea (mean respiratory rate >2 SD above normal OR mechanical ventilation) and (4) leucocyte count (elevated or depressed).31 ,32 Sepsis was defined as SIRS plus positive blood culture. Prolonged antibiotic administration was defined as intravenous administration of any antibiotic for >48 h.

Subject clinical variables, sample level stacked bar plots of the most common taxonomical units and comparison of Shannon diversity indices by clinical variable. (A) Subject table of clinical variables. BW, birth weight, grams; EA, early antibiotics, days; GA, gestational age, weeks; PPROM, preterm premature rupture of membranes; SDI, Simpson diversity index at the sample level. (B) Sample level stacked bar plots. The legend shows the colour scheme for the six most abundant genera. Samples are grouped by clinical outcome and ordered sequentially for each patient. Three of the patients show samples with high abundance of Staphylococcus. Patients 2 and 3 show unusually high abundance of Veillonella and Clostridium when compared with the rest of the patients. (C) Patients comparisons based on patient subgroups. p Values are derived from pairwise t-tests, red asterisks indicate statistical significance based on 0.05 threshold. Box plots show the minimum, quartiles, median and maximum SDI values.
Antibiotic exposure
Maternal antibiotic exposure, and complete neonatal antibiotic exposure data was collected and analysed relative to changes in the microbiome over time.
Sample collection
Serial stool samples were collected weekly (routine) or at sepsis evaluation, beginning with the first stool; the day of life at which first meconium was passed ranged from day 3 to day 14 (figure 1A, meconium). Stool samples were collected using sterile wooden depressors and transferred into sterile tubes, then aliquotted in a laminar flow hood and stored at −80°C until processing. Samples were collected for the subjects until discharge or transfer to another institution (for a total of 8 to 12 weeks) and analysis for this study encompassed samples from the first three stools to evaluate first episode of late-onset sepsis.
Sample processing
DNA was extracted using the MoBio Powersoil bacterial DNA isolation kit. 50 mg of stool sample was dissolved in phosphate-buffered saline, then DNA was extracted according to manufacturer's instructions. The DNA was used to construct PCR amplicon libraries for parallel sequencing.
Massively parallel sequencing
Massively parallel 454 pyrosequencing was performed at the Josephine Bay Paul Marine Biological Laboratories. Pyrosequencing was targeted at the 16S rRNA gene using titanium amplicon sequencing. A cocktail of five fused primers at the 5′ end of the V6 region and four primers at the 3′ end are designed to capture the full diversity of rRNA sequences represented in molecular databases.26 ,33 Taxonomic identifiers were assigned to pyrotags by using the rRNA indexing algorithm Global Assignment of Sequence Taxonomy (GAST),24 which compares pyrotags to known rRNA genes that have already been placed in a phylogenetic framework of more than 1 000 000 nearly full-length rRNA reference sequences (RefSSU) on the SILVA database.34 The tag-mapping methodology GAST for taxonomic assignments of environmental V6 Pyrotags and the V6 reference database are freely available through the Visualisation and Analysis of Microbial Population Structure website http://vamps.mbl.edu/resources/faq.php#gasting. All new data generated from sequencing has been deposited in the (National Centre for Biotechnology Information of the National Institutes of Health and the US National Library of Medicine) GenBank (SRA) with accession number SRA043957.1.
Data analysis
Faecal microbiota of the infants was compared between all subject subsets defined by clinical data (figure 1A). Operational taxonomical units defined at a 3% sequence difference cutoff were treated as the terminal taxonomical rank. Richness was defined as the total number of taxonomic units. Diversity was evaluated using the Simpson diversity index (SDI), which represents the sum of the relative proportion of bacterial species (1 – D; D = sum (n × (n – 1)/N/(N – 1)) where n=# organisms/species, N=# organisms; 1=infinite diversity. Sample distances were computed using the Jaccard distance. Statistical analysis and generation of graphical output were performed using the R statistical language.
Results
Patient cohort
Subjects were VLBW infants delivered at 24–27-weeks gestation with birth weight ranging from 510 to 1080 g (figure 1A). Three subjects' maternal complications included PPROM, three had chorioamnionitis (placental pathology) and five were delivered by caesarean section. All subjects were initially fed maternal breast milk, and all subjects eventually received bovine human milk fortifier added to breast milk. None of the subjects had culture-proven early-onset sepsis (sepsis diagnosed <72 h of life). The subjects observed in the study demonstrated three distinct clinical patterns. First, two subjects developed late-onset sepsis, with blood cultures positive for C albicans and S aureus (subject 1) and S marcescens (subject 4; figure 1A) (sepsis group) during the week of life after sample 3 was collected. Second, two subjects (subjects 5 and 6) exhibited early symptoms consistent with SIRS (figure 1A, SIRS). Finally, two subjects did not develop sepsis (subjects 2 and 3; figure 1A, healthy).
Antibiotic exposure
Maternal antibiotic exposure during the course of labour included the following: no antibiotics for subjects 1 and 5, and one dose (partial treatment) <4 h before delivery for subjects 2, 3, 4 and 6. Subjects all received broad-spectrum antibiotics for a minimum of the first 2 days of life (ampicillin and gentamicin). Four of six subjects received prolonged, empiric antibiotic coverage secondary to clinical instability at the time of birth that persisted (figure 1, table A, early antibiotics). Subjects 1 and 4 who later developed late-onset sepsis following collection of sample 3, were treated as follows: Subject 1 with ampicillin and gentamicin for 3 days, then with clinical decompensation on day 4, blood cultures were resent and antibiotics were changed to vancomycin and cefotaxime for an additional 7-day course. Subject 4 received ampicillin and gentamicin for 7 days for presumed sepsis, and then demonstrated gastrointestinal symptoms concerning for necrotising enterocolitis, and antibiotics were changed to vancomycin and cefotaxime on day 7 for a further 5 days. Subjects 5 and 6 both received prolonged, empiric antibiotics (ampicillin and gentamicin) for 6 and 7 days, respectively for presumed culture-negative sepsis (SIRS).
Dominant genera
324 350 sequences were obtained from serial samples. Ninety-five per cent of the sequences observed were represented by eight genera (figure 1B). Most subjects receiving prolonged antibiotics (subjects 1, 4 and 6) had a preponderance of Staphylococcus in at least one sample (figure 1B). Subject 1 had a predominance of Staphylococcus species at birth and went on to develop S aureus sepsis in week 4. Subjects 2 and 3 were considered healthy subjects in the study and these subjects had a predominance of Enterobacteriaceae and Gammaproteobacteria, with significantly higher representation of Firmicutes, including Clostridium and Veillonella, bacteria that were not dominant in subjects who developed sepsis or were exposed to prolonged antibiotics (figure 1B).
Microbial diversity
Overall diversity (SDI) for combined samples for each subject demonstrates patterns related to clinical course (figure 1C). Subjects 1 and 4 who ultimately develop sepsis had decreased diversity compared to other subjects beginning with birth (figures 1B, C, sepsis). SDI was statistically significantly decreased for subjects whose mothers had chorioamnionitis (diagnosed by placental pathology), subjects who were exposed to prolonged empiric antibiotics beginning at birth, and for subjects who develop late-onset sepsis (figure 1C). Subjects who have a healthy clinical course despite their prematurity demonstrate increased diversity from birth in meconium stools (figure 1B), and continue to demonstrate increased diversity throughout all samples. Thus, diversity of the microbiota does appear to track with the development of sepsis.
Meconium, risk and healthy sample clusters
Our analysis indicates that the meconium of all subjects was not sterile and had a predominance of Enterobacteriaceae and Staphylococcus. Three statistically significant clusters emerged from the data as shown in figure 2A: (1) a cluster containing all but one meconium sample (M cluster), (2) a cluster containing at least one sample from three of the high-risk patients (R cluster) and (3) a cluster containing all healthy patient samples (H cluster). Surprisingly, samples from the same patient did not cluster together indicating that the samples are more similar between patients than within patients. Instead, the clustering of the samples appears to be driven by the clinical state of the patients (figure 2A). Having at least one sample that belongs to the R cluster is indicative of the final clinical outcome (figure 2A). The SDI for the H cluster is statistically significantly different from both the M and R clusters (figure 2B).

Clustered heat map demonstrating clusters of meconium (M) genera, risk for sepsis (R) genera and healthy genera (H). (A) Clustered heat map. Data are filtered based on abundance across samples, normalised and base ten log transformed. Similarity between genera based on their abundance across samples is computed with pvclust. Colours are assigned to genera in taxonomical order. Three significant and disjoint clusters appear at the 0.05 significance level, indicated with red bars. These are named eM (enriched in meconium samples), eH (enriched in healthy samples) and dH (depleted from healthy samples). Similarity between samples is computed with pvclust. SDI bar plots are shown in gray to the right of the sample names. Samples are coloured by patient type: red – sepsis, orange – systemic inflammatory response syndrome, green – healthy; with the exception of meconium samples which are blue. Three significant clusters appear at the 0.05 significance level. These are named M (meconium), R (risk), H (healthy) based on their sample composition. The R cluster shows a marked decrease in abundance for all genera and a signature overabundance of Staphylococcus. (B) SDI box plot for the three sample based clusters. Box plots show the minimum, quartiles, median and maximum SDI values.
Meaningful bacterial clusters
Significant clusters also emerged at the genus level (heat map, figure 2A). Of the three disjointed clusters, the large cluster contains the genera that are enriched in the M sample cluster (eM genera cluster), the middle cluster contains the genera enriched in the H sample cluster (eH genera cluster) and the small cluster contains the genera depleted in the H sample cluster (dH genera cluster). The only genus enriched in the R sample cluster is Staphylococcus. This correspondence highlights the signature genera for each of these three clinical states.
Characterising the transitions between stable states
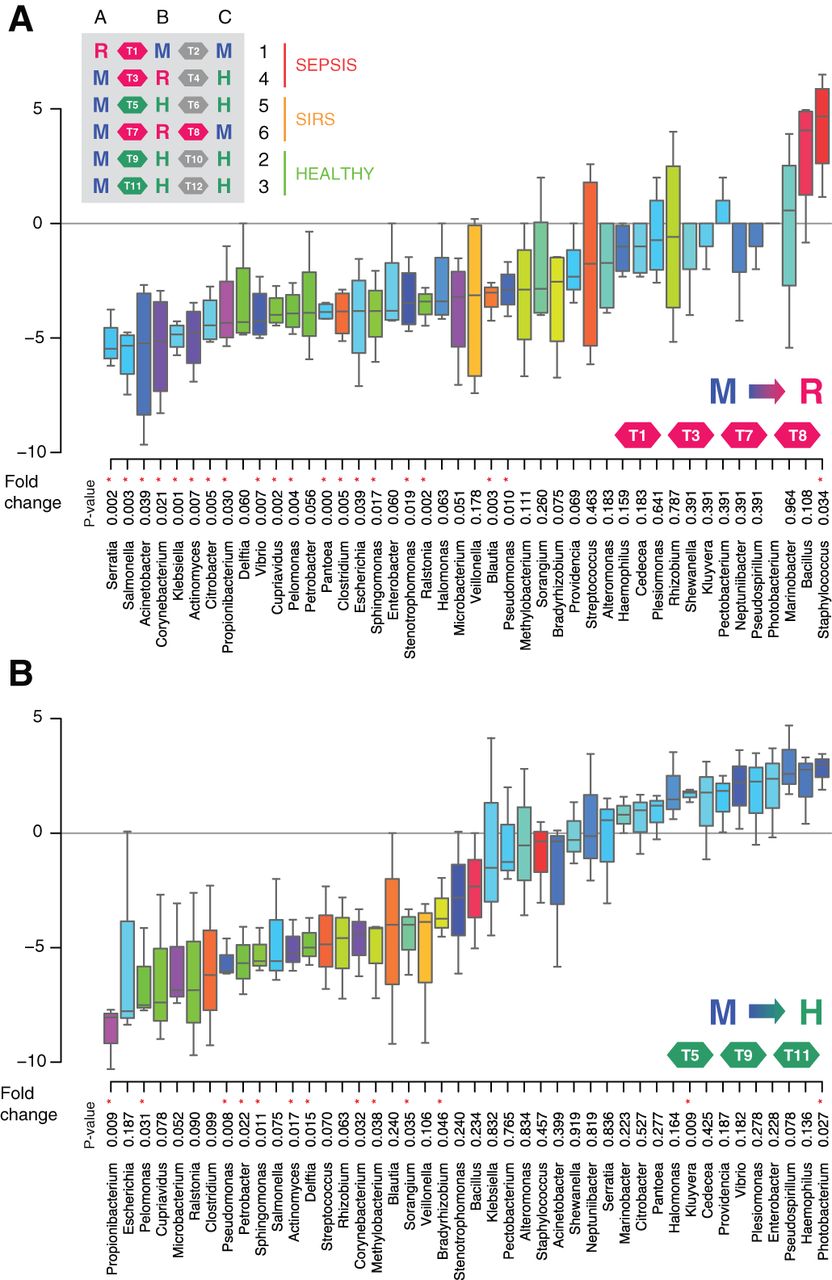
The consistency of patterns between samples from subjects suggests the microbiome in development is attracted towards stable states. In order to characterise the transitions between the three states discovered, we calculated fold changes across genera for each of the three transitions: M to H, M to R and H to R. The transitions from M to R states revealed the consistent decrease of 26 genera, with Staphylococcus as the only consistently increased genus (figure 3A). Despite both state transitions showing an overwhelming majority of decreasing genera (figure 3A,B), the M to H transition is characterised by an increase in diversity, and the M to R transition in a decrease of diversity.

{kind=link}
{kind=link}
{kind=link}
Genera transition plots between the M (meconium) state and the R (risk for sepsis) and H (healthy) states. (A) Genera transition plot for the M to R state transition. Inset table shows all 12 transitions within the 18 samples. Fold-changes displayed as box plots are derived from four transitions: T1, T3, T7 and T8. p Values are based on t-tests. Asterisks highlight significance at the 0.05 level. Staphylococcus is the only significantly enriched genus when transitioning to the R cluster. (B) Genera transition plot for the M to H state transition. Fold-changes displayed as box plots are derived from three transitions: T5, T9 and T11. Kluyvera and Photobacterium are the only significantly enriched genera when transitioning to the H cluster. Data are filtered based on abundance across samples, normalised and base two log transformed.
Discussion
Our indepth study in a limited cohort, using massively parallel pyrosequencing technology, outlines the developing microbiome in serial longitudinal samples from premature infants who are highly susceptible to infection, and identifies patterns as they relate to health and disease. Limitations of our study include the small sample size; therefore inference with regard to causality is limited. However, despite our small sample size, our findings support those of Arumugam35 as we identified distinct clusters of microbial genomic information or ‘enterotypes’ that were driven by species composition, and correlated with clinical outcomes. Overall, we were able to lay the groundwork for larger studies to better characterise the developing microbiome in premature infants at risk for sepsis.
We demonstrate that microbial patterns in meconium were similar, but were less diverse beginning from birth in high-risk infants who develop sepsis. Until recently, meconium was thought to be sterile.36 ,37 Recent studies have demonstrated and corroborated our findings that meconium is not sterile in this population suggesting an intrauterine origin of bacteria.37 Of note, VLBW infants such as those in this study, often have delayed passage of stools, and microbes present in meconium may reflect intrauterine exposures (intrauterine infection/colonisation secondary to PPPROM, or maternal antibiotics), delivery exposures, or antimicrobial exposure and interventions in the neonatal intensive care unit (NICU).
Our results demonstrate that microbial diversity was affected by history of maternal chorioamnionitis before delivery, with persistence of significantly lower microbial diversity. Interestingly, the mothers in this cohort were not exposed to significant amounts of peripartum antibiotics secondary to the urgency of their deliveries. These infants were perhaps in an unsterile environment inutero, and we postulate that the lower diversity was potentially attributable to maternal antibiotic exposure or an overabundance of primary pathogens. DiGiulio et al demonstrated in prenatal amniocentesis evaluation in the face of preterm labour that microbial density showed a dose relationship between microbial DNA and likelihood of premature delivery.38 Investigation of intrauterine environment, including detailed analysis of maternal infection and antibiotic exposure, and subsequent neonatal colonisation patterns in a large cohort might identify changes in colonisation that have clinical implications. We also demonstrate shifts in microbial populations attributable to antibiotic exposure; the associated decreased diversity may result in lengthening the instability of the developing microbiota and increased risk of disease in premature infants. Similar to our findings but in adults, Dethlefsen found profound alteration in intestinal microbial populations following ciprofloxacin, with incomplete recovery over a period of 10 months.18 Ubeda et al identified in humans that domination by vancomycin-resistant enterococcus preceded bloodstream infection in immune-compromised adults.23 We theorise that, similar to Ubeda's findings, pathogens are selected for following broad-spectrum antibiotic administration in premature infants. The complex interactions between the nearly blank canvas of the neonatal intestine, colonising microbes, nutritional and antibiotic practices, the developing immune system and risk of infection, may be significantly influenced by the intensive care environment and interventions required for premature infants.8 ,9 ,39
We have correlated microbial colonisation in high-risk preterm infants with disease risk, and potential future implications include altering antibiotic regimens or enlisting the targeted use of probiotics. Wang et al demonstrated a decreased microbial diversity in subjects who developed necrotising enterocolitis, and this group was also exposed to more antibiotics than controls.40 These findings were similar to our septic subjects who had overall decreased diversity, beginning with meconium stools and persisting until the onset of sepsis. We also demonstrate colonisation patterns that seem specific to subjects who develop sepsis, with a Proteobacteria and Firmicutes (Staphylococcus) predominance, whereas healthy subjects who receive limited antibiotics (<3 days total) and did not develop sepsis ultimately demonstrate an increase in relative abundance of anaerobes, potentially mirroring more ‘mature’ microbial communities, including Clostridium, Klebsiella and Veillonella.2 Jacquot et al identified a Staphylococcus predominance in the most premature infants studied, and found the least diversity of bacteria in the most premature infants.41 Staphylococci have been described as commensals but are also among a short list of ‘translocating’ bacteria when present in abundance, which may be a contributing factor to the common risk of Staphylococcal sepsis in the NICU environment.2 ,42 ,43 Interestingly, the majority of contemporary studies of the microbiome using non-culture-based techniques are in high-risk neonatal populations rather than healthy term infants. The studies that have been published in healthy infants describe different results, including a Proteobacteria and Bifidobacterium predominance using targeted small subunit (SSU) rRNA rRNA44 mirroring the traditional culture-based studies, and Palmer et al, using microarray, describes a predominance of Proteobacteria, Firmicutes and Bacteroidetes45 which mirrors our findings for infants at risk for sepsis, with Proteobacteria and Firmicutes (Staphylococcus) predominance.
Conclusions
The microbial patterns of meconium were similar among all premature infants, but less diverse from birth in infants who ultimately develop late-onset sepsis. We noted a divergence occurring between healthy infants and those who subsequently developed sepsis, with Staphylococcus predominance in subjects who go on to develop late-onset sepsis. Empiric, prolonged broad-spectrum antibiotics profoundly decreased gut microbial diversity and promoted a pathogen-predominant microbiota that was associated with sepsis due to the predominant pathogen in the microbiome. Our data suggest that premature infants who do not develop neonatal sepsis may have a definable ‘healthy microbiome’ that larger prospective studies might further elucidate.
Acknowledgments
The authors would like to thank the infants and families from the intensive care nursery who participated in the study as well as the dedicated nurses and unit team who assisted with the study. The authors would like to acknowledge Dr Salvador Almagro-Moreno for his insightful thoughts on information design, and Ms Nisha Jambulingam for her work with sample processing. The authors also deeply appreciate the leadership and knowledge provided by Ms Molly Housman of the Dartmouth Hitchcock Division of Infectious Diseases and Dr Hilary Morrison of the Josephine Bay Paul Center Marine Biological Laboratories. The authors would also like to thank Dr Bruce Stanton for his mentorship, and Dr Doug Hill and Tom Caldwell for their significant technical expertise.
References
Footnotes
-
Funding The Hearst Foundation, The Synergy Grant (Dartmouth), The Joshua Burnett Career Development Award through the Hitchcock Foundation (Dartmouth), the Department of Pediatrics, Dartmouth. This work was also supported by a pilot grant from the Cystic Fibrosis Foundation Research Development Program (STANTO07R0). RCS and JHM were funded by NIH R01 AI59694. JAF was funded in part by NIH grants P20RR16448 and P20RR016454 and by NSF DBI0939454.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval Dartmouth College CPHS IRB.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement The authors have provided open access to their sequencing data to other scientists by publishing the sequencing data in the following location: all new data generated from sequencing has been deposited in NCBI GenBank (SRA) with accession number SRA043957.1.