Article Text

Abstract
Insulin is synthesised, stored, and secreted from pancreatic β cells. These are located within the islets of Langerhans, which are distributed throughout the pancreas. Less than 2% of the total pancreas is devoted to an endocrine function. When the mechanisms that control insulin release are compromised, potentially lethal diseases such as diabetes and neonatal hypoglycaemia are manifest. This article reviews the physiology of insulin release and illustrates how defects in these processes will result in the pathophysiology of hyperinsulinism of infancy.
- hypoglycaemia
- ATP sensitive potassium channels
- insulin secretion
- nesidioblastosis
- hyperinsulinism
- diazoxide
Statistics from Altmetric.com
- hypoglycaemia
- ATP sensitive potassium channels
- insulin secretion
- nesidioblastosis
- hyperinsulinism
- diazoxide
Pancreatic β cells have evolved to release insulin in response to a host of different physiological regulators: from neurotransmitters and neuropeptides, to circulating hormones, amino acids and, of course, glucose. Each of these molecules reaches the β cell by a number of fundamentally different routes. A dense network of blood vessels keeps the islets of Langerhans informed about the nutrient and hormonal status of the blood. The islets are also richly innervated by an array of neural influences that can both inhibit and stimulate insulin release. Paracrine modulation is also important. Within the islets, somatostatin secretion from δ cells will inhibit insulin release, whereas glucagon from α cells will stimulate insulin secretion. Finally, β cells are also influenced by “autoregulatory events”. Insulin—for example, which has long been thought to have no effect on secretion or to have an inhibitory influence, has recently been shown to act upon β cells through its own receptor to enhance both insulin biosynthesis and secretion.1-3 When faced with such a multitude of diverse signalling molecules, the decoding of signalling information by the cell becomes crucial. Recent progress in the area of “stimulus–response coupling” has provided new insights into the control of β cell operation and evidence that defective processes can lead to the manifestation of disease.
In the hierarchy of insulin secretagogues, glucose is the prevailing influence. At concentrations that elicit insulin release, glucose has pronounced effects upon several aspects of β cell function. These culminate in both “first phase” and “second phase” secretion, and the subcellular mechanisms that control these processes are in the process of being elucidated.
The electric β cell
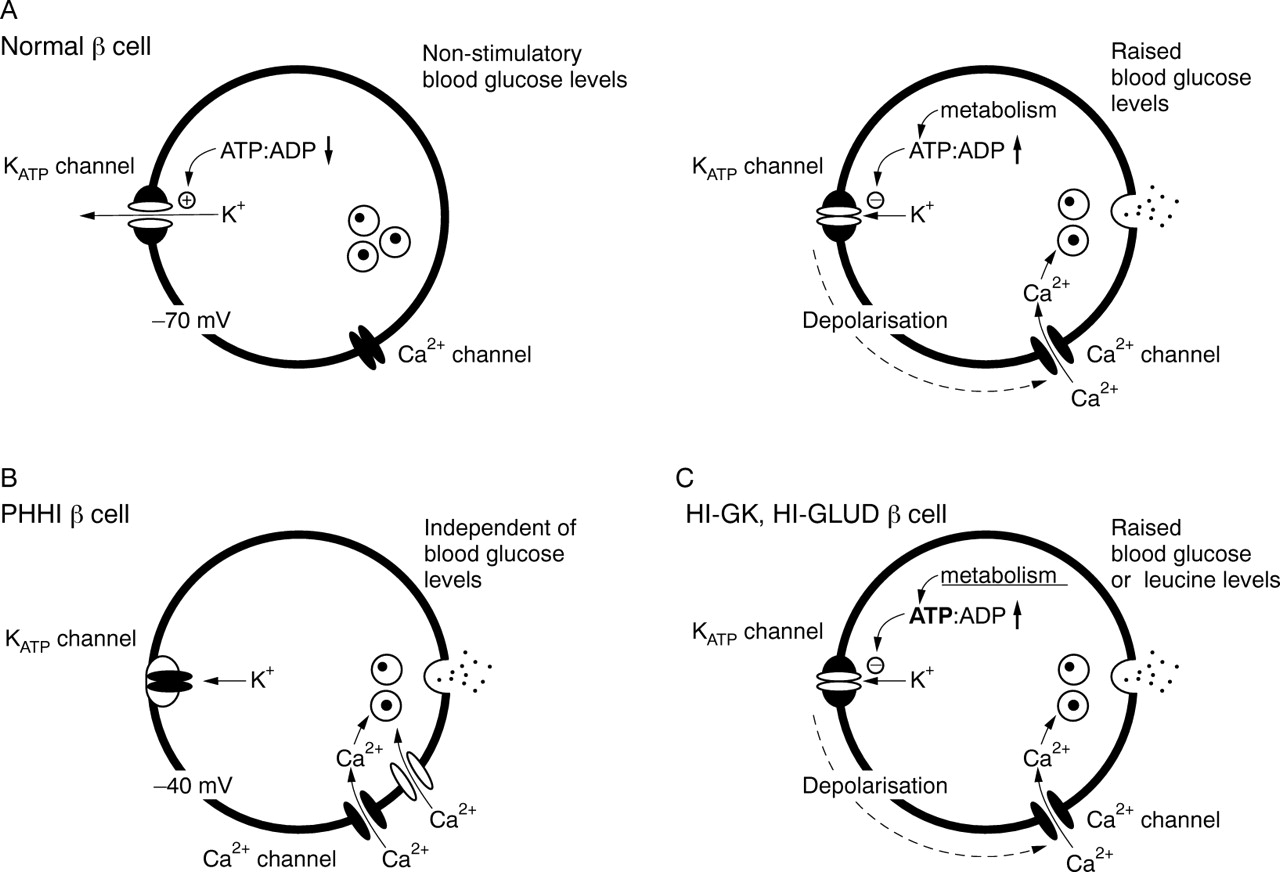
β Cells are electrically active. In the presence of stimulatory concentrations of glucose (typically greater than 5.5 mM) they release insulin in a pulsatile manner. Oscillations in insulin secretion are dependent upon the concentration of glucose: increasing in frequency with increasing concentrations of glucose. These events are underpinned by cycles of change in the cell membrane potential, which in turn generates oscillations in the intracellular free calcium concentration. Metabolism of glucose is crucial to initiate these changes in the membrane potential. This is facilitated through the closure of potassium ion channels at the plasma membrane—the ATP sensitive K+ (KATP) channels. Ion channels are plasma membrane “tunnels”, and under resting conditions the membrane potential in β cells is determined by the Na+–K+ ATPase pump and K+“leak” through open KATP channels. After glucose uptake and metabolism by both glucokinase (the “glucose sensor”) and mitochondrial events, KATP channels are closed as a consequence of the increased intracellular ATP/ADP ratio. KATP channel closure leads to a depolarisation of the cell and the opening of voltage dependent Ca2+ channels. The subsequent influx of Ca2+ down a > 10 000-fold concentration gradient, leads to a sharp rise in the cytosolic Ca2+ concentration close to the membrane, which then initiates the release of insulin by the process of exocytosis (fig 1). In this manner, KATP channels control the “set point” of β cell electrical activity, and their modulation clearly has a direct bearing on the regulated release of insulin. This has clinical implications. Thus, the selective inhibition of KATPchannels with drugs such as the antidiabetic sulphonylureas (for example, glibenclamide and tolbutamide) will mimic the actions of glucose and promote insulin release. Conversely, KATPchannel “openers”, such as the hyperglycaemia inducing compound diazoxide, will exert the opposite effect and inhibit secretion by preventing voltage dependent Ca2+ entry.4Direct inhibition of voltage gated Ca2+ channels with antagonists, such as verapamil and nifedipine, will similarly inhibit secretion from β cells.5

The ionic control of insulin release from pancreatic β cells. (A) In normal β cells, the resting membrane potential (approximately −70 mV) is determined by open KATPchannels. When the extracellular glucose concentration is raised, glucose is taken up by the β cell and glucose metabolism is initiated. The rate limiting step in this process is glucokinase and the formation of glucose-6-phosphate. Subsequent metabolic events lead to an increase in the cytosolic ATP/ADP ratio, and the closure of KATP channels follows. This leads to membrane depolarisation and the opening of voltage dependent Ca2+channels. Increased Ca2+ influx then initiates the release of insulin through exocytosis of secretory granules. (B) Persistent hyperinsulinaemic hypoglycaemia of infancy (PHHI) arises from defects in either the KIR6.2 or SUR1 genes. Because these β cells lack operational KATP channels, the membrane potential is spontaneously depolarised (approximately −40 mV) in the absence of glucose metabolism. This leads to the persistent activation of Ca2+ channels, causing unregulated entry of Ca2+ and persistent release of insulin as a consequence. (C) Hyperinsulinism of infancy (HI) can arise from an acquired loss of KATP channel function. HI-GK and HI-GLUD are not caused by KATP channel defects, but by mutations in the genes encoding either glucokinase (GK) or glutamate dehydrogenase (GLUD). These defects augment glucose and leucine metabolism in the β cell, lead to excessive ATP production, and the enforced closure of KATP channels. This is thought to cause inappropriate membrane depolarisation and unregulated Ca2+ entry.
Controlling the Ca2+ signal
In β cells (as in all cell systems) entry of Ca2+ across the plasma membrane (Ca2+ influx) is only one of the processes that govern intracellular Ca2+homeostasis (fig 2). Such is the dominance of Ca2+ as an obligatory signalling molecule, that inside β cells the concentration of Ca2+ is buffered to diminishingly small values. This is maintained by “extrusion” of Ca2+ across the plasma membrane and “sequestration” of Ca2+ into internal Ca2+ storage organelles by the energy requiring Ca2+ ATPases—plasma membrane Ca2+ ATPases (PMCA) and sarcoendoplasmic reticulum Ca2+ ATPases (SERCA), respectively. The activity of these pumps is exquisitely coordinated with mechanisms responsible for Ca2+ influx (ion channels) and Ca2+ release (mobilisation) from intracellular stores. This mainly occurs through inositol (1,4,5) trisphosphate receptor gated Ca2+ channels, but there is also evidence that ryanodine receptor mediated events might be involved. In addition, β cells have developed a direct mechanism to aid in the refilling of internal stores in the presence of maintained stimulation. The concentrations of Ca2+ contained within sarcoendoplasmic reticulum structures mainly govern “capacitive Ca2+entry”. As yet, we have an incomplete understanding of how the communication is established and how it is regulated in β cells. Candidate genes such as TRP and TRPL, and “Ca2+ release activated non-selective ion channels (ICRAN)” appear to be involved. It is thought that after depletion of Ca2+from internal stores, Na+ entry through these channels leads to a depolarisation of the cell membrane potential and the activation of voltage gated Ca2+ channels.6 ,7What is not clear is how the internal Ca2+ stores are able to signal to the cell membrane, nor how this is regulated.

Intracellular Ca2+ homeostasis in pancreatic β cells. The regulation of Ca2+ mediated insulin release is governed by: (1) Ca2+ efflux controlled by plasma membrane Ca2+-ATPases (PMCA); (2) Ca2+ sequestration by sarcoendoplasmic reticulum Ca2+-ATPases (SERCA); (3) mobilisation from internal stores; and (4) Ca2+ entry through voltage gated Ca2+ channels and capacitive Ca2+ entry. Capacitive Ca2+ entry is linked to internal store content by the activity of non-selective cation channels (ICRAN). Once open, Na+ influx causes a depolarisation of the cell and the activation of voltage gated Ca2+ channels. ACh, acetylcholine; CCK, cholecystokinin; DAG, diacylglycerol; ER, endoplasmic reticulum; I(1,4,5)P3, inositol (1,4,5) trisphosphate; PIP2, phosphatidylinositol (3,4,5) bisphosphate; PLC, phospholipase C.
Regulating insulin release
Glucose metabolism coupled to KATP channel activity governs first phase insulin release: the secretion—by exocytosis—of preprepared insulin containing granules. If we use the information established in yeast and neuronal systems for ongoing studies in the β cell, it is possible to predict the distal steps associated with the regulated release of insulin. Upon β cell activation, the granules have to be translocated to the plasma membrane. Even though some granules might be present at the membrane and available for exocytosis before cell activation, sustained secretion requires that translocation occurs as a regulated process. After this, the granules must dock; they are then primed for the final event, which is fusion of the granule membrane with the plasma membrane, followed by exocytosis. Subsequently, endocytosis must succeed exocytosis to complete the cycle and allow the cell to continue to secrete insulin for lengthy periods. Without doubt, this simple classification of the late stages of stimulus–secretion coupling understates the complexity of the overall mechanism. The sequence of reactions leading to insulin release will involve many interacting moieties and different isoforms in keeping with the “SNARE (SNAP receptor) hypothesis”. Figure 3 illustrates a minimal description of the components of this process in β cells (recently reviewed by Lang8).
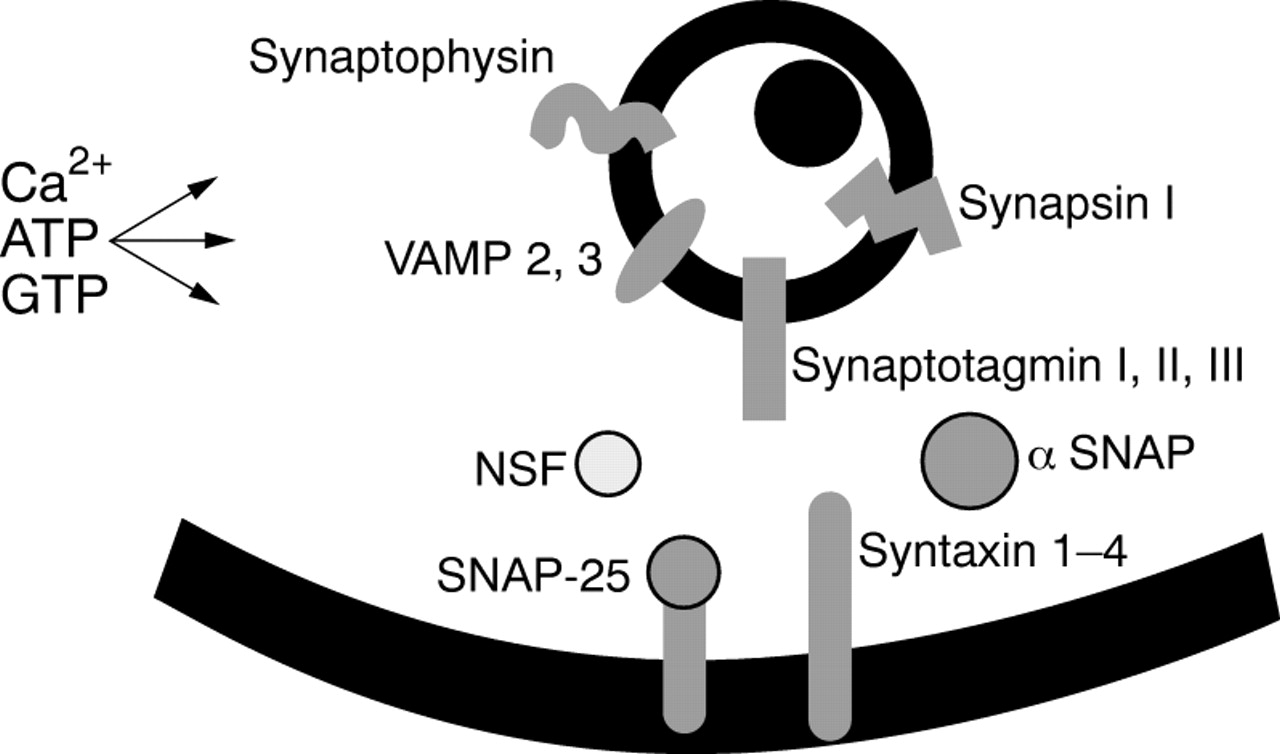
The components of regulated exocytosis in pancreatic β cells. Multiple components control the late stages of stimulus–secretion coupling and exocytosis. These include the granule associated proteins synapsin 1, synaptophysin, VAMP (vesicle associated membrane protein)/synaptobrevin and synaptotagmin. NSF (N-ethylmaleimide sensitive factor) and SNAPs (soluble NSF attachment proteins) are located in the cytosol, whereas syntaxin and SNAP-25 are found on the plasma membrane. For docking of the secretory granule to occur, the VAMP/synaptobrevin (v-SNARE) in the granule membrane binds to syntaxin and SNAP-25 (these two make up “t-SNARE”) in the plasma membrane. Subsequently, NSF and SNAPs complete the formation of the fusion complex that leads finally to exocytosis.
A second important mechanism in the control of glucose dependent insulin secretion has been documented as the “KATPchannel independent pathway” of glucose action.9 This pathway leads to “augmented” insulin release in the presence of raised cytosolic Ca2+ concentrations (raised by the KATP channel dependent pathway of glucose signalling) (fig4). Experimentally, the pathway can be demonstrated by causing Ca2+ entry under conditions that prevent KATPchannel operation—for example, with diazoxide (which pharmacologically activates the KATP channels) or sulphonylureas (which inhibit KATP channels).9-14 With the KATP channels clamped, glucose induces a concentration dependent augmentation of insulin secretion, which is responsible for second phase insulin release. The mechanisms that govern this effect are largely unresolved. However, it is recognised that glucose metabolism is fundamentally required, and that the site of action is distal to the rise in intracellular Ca2+. As an additional level of complexity, KATP channel independent actions of glucose to augment insulin release have also been shown to occur independently of raised cytosolic Ca2+.15 This glucose augmentation pathway involves the simultaneous activation of protein kinases A and C (as would occur during exposure to hormones, neuropeptides, and neurotransmitters) and is dependent upon ATP and GTP availability to fuel the process of exocytosis.
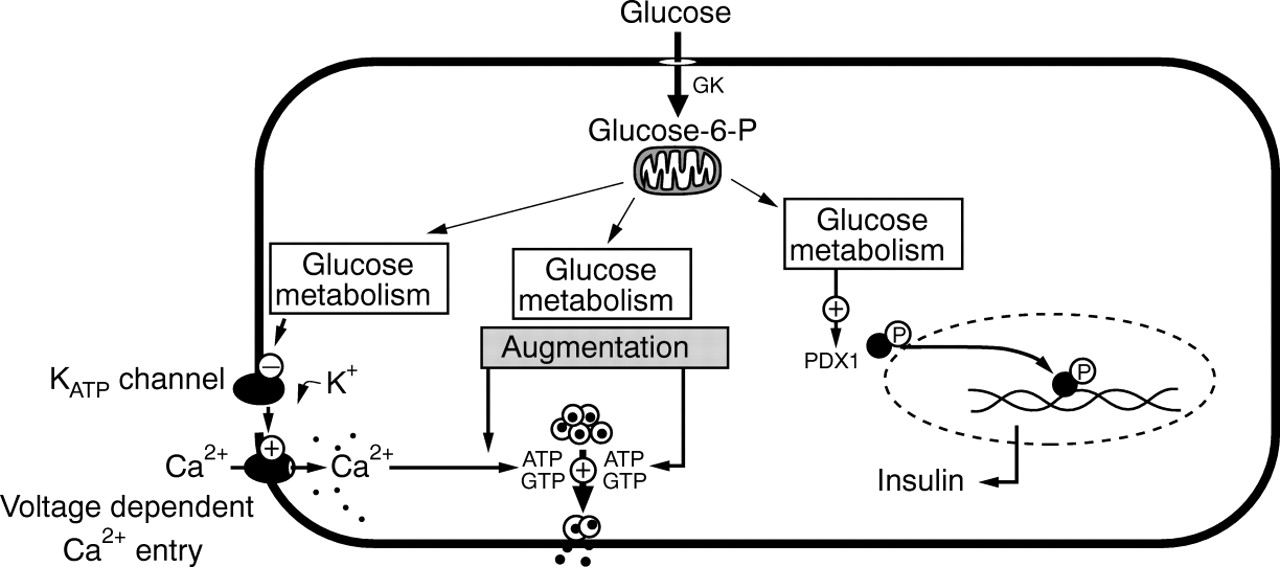
The control of insulin release from pancreatic β cells. Glucose metabolism is responsible for: (1) KATPchannel dependent Ca2+ influx leading to insulin release; (2) KATP channel independent pathways of glucose augmentation; and (3) insulin biosynthesis.
Replenishing the reserves of insulin
Concomitant with the control of second phase insulin secretion, glucose also stimulates the production of new insulin molecules by the translation of preformed mRNA encoding insulin (within minutes), and over longer periods of time (hours) by increased transcription of the insulin gene. One key element in linking cytosolic events to nuclear signalling is the homeodomain transcription factor PDX1.16Glucose metabolism causes phosphorylation of PDX1 and translocation of the transcription factor to the nucleus. In addition to insulin, PDX1 also binds to the promoter regions of a number of other genes preferentially expressed in the β cell, including those encoding glucose transporter GLUT-2, glucokinase, and islet amyloid polypeptide. Because PDX1 is also an important factor determining lineage in β cell development,17 it is not surprising that defects in the PDX1 gene cause maturity onset diabetes in the young type 4 and pancreatic agenesis. The role of PDX1, or indeed other similar molecules, in hyperinsulinism of infancy (HI) has not been critically examined. However, it is interesting to note that impaired PDX1 activity is a phenotypic feature of the persistent hyperinsulinaemic hypoglycaemia of infancy (PHHI) derived human β cell line, NES2Y (see below).18 Whether this is a primary defect, or a consequence of the loss of KATP channels in the β cell has yet to be determined.
Controlling the coordinated release of insulin from β cells in response to physiological agents is clearly a highly integrated but ordered process. Recent advances in the field of β cell physiology have provided new insights into these processes. Equally important have been advances in the area of the pathogenesis of insulin release. One of the most compelling examples of the integration of physiology with pathophysiology has been the revelation that HI caused by PHHI is a KATP channelopathy.
KATP channel structure and regulation
β Cells express a KATP channel complex formed by subunits belonging to at least two distinct families of proteins (recently reviewed19 ,20) (fig 5). The K+selective pore is formed by the Kir6.2 subunit, a member of the inward rectifier K+ channel family. Kir6.2 comprises 390 amino acids, and has a predicted membrane topology, with two α-helical transmembrane domains linked by a highly conserved sequence of amino acids. Because this linking region shares sequence homology with the P or H5 region of voltage gated K+ channels, it is thought to be important for the control of K+ selectivity through the pore (fig 5). The other subunit, a larger protein, is a receptor with high affinity for sulphonylureas, designated SUR1.21 Two closely related genes encode two sulphonylurea receptors, SUR1 and SUR2.22 Three splice variants of the SUR2 gene have been described and designated, SUR2A, SUR2B, and SUR2C.19 Human SUR1 is 1581 amino acids in length and like the cystic fibrosis transmembrane conductance regulator and the multidrug resistance protein it is a member of the superfamily of ATP binding cassette proteins. SUR1 has 17 predicted transmembrane domains (TMDs)23 organised into three regions (TMD0, TMDI, and TMDII), and two intracellularly disposed nucleotide binding domains (NBDs) (fig 5). Neither the native Kir6.2 subunit nor the SUR1 subunit will form operational K+ channels independently. However, when coexpressed, K+ channel currents are generated that closely resemble those of the native β cell KATP channel complex.24 ,25 KATP channels in other tissues are heteromultimeric complexes of different Kir6.x and SUR proteins—for example, cardiac muscle: Kir6.2 + SUR2A; smooth muscle: Kir6.2 + SUR2B; and the smooth muscle nucleotide activated channel: Kir6.1 + SUR2B.19

Components of KATP channels in pancreatic β cells. (A) The predicted topology of the K+ channel subunit, SUR1, an ATP binding cassette protein. Note the characteristically high number of transmembrane spanning domains 1–17 and the two intracellularly disposed nucleotide binding domains (NBDs). Also shown are the predicted sites for potassium channel opener binding (KCOI, KCOII) and sulphonylurea binding (SURB). Note that these are located towards the C-terminal region of the molecule. Kir6.2 has two transmembrane domains and an inner loop that controls K+influx. (B) The heteromultimeric KATP channel complex is an obligatory octameric structure composed of (Kir6.2/SUR1)4. Kir6.2 is located toward the centre of the complex.
SUR1, Kir6.2, and regulation of KATP channels
The topological organisation of the KATP channel is thought to be that of an obligatory octameric complex, formed by four Kir6.2 subunits lining the pore, coupled to four SUR1 subunits—(SUR1 + Kir6.2)4 (fig 5B).26 ,27Through molecular biology techniques, recent advances have been made in determining what contribution each of the subunits makes to the properties of the channel complex. Kir6.2 determines the biophysical properties, such as the correct ion selectivity, rectification and “gating” properties of the complex.19 ,20 The sensitivity of KATP channels to physiological regulators, such as the adenine (ATP, ADP) and guanosine (GTP, GDP) nucleotides, is a complex process involving both subunits. Kir6.2 is thought to be the major site for ATP binding and ATP induced channel closure, whereas ADP (and GDP) interacts with one of the nucleotide binding domains of SUR1.19 ,20 ,28 In this manner, both subunits are important for ATP/ADP mediated KATP channel activity, with the actions of ADP on SUR1 antagonising the effects of ATP at Kir6.2. SUR1 influences the trafficking and distribution of Kir6.2, but both subunits contribute to the ordered assembly and location of the complex at the plasma membrane.29 ,30 The protein kinase A induced phosphorylation of KATP channels, as would occur during stimulation of the β cell with receptor mediated events such as glucagon-like protein, gastric inhibitory peptide, and pituitary adenylate cyclase activating peptide (GLP-1, GIP, and PACAP), also involves both SUR1 and Kir6.2.31
Pharmacological regulation of KATP channels is also governed by interactions with each of the subunits, but interestingly different classes of agents appear to regulate differentially either SUR1 or Kir6.2. Antidiabetic sulphonylureas have a low affinity binding site on Kir6.2, but bind with high affinity to SUR1.32 ,33The sulphonylurea receptor binding site (SURB) is currently thought to lie between the transmembrane sequences 14–16 (residues 1035–1277)32 in the TMDII part of the molecule (fig 5B), and occupancy of just one of the four SURBs in each complex is enough to induce channel closure.34 The identification of SURB at this particular site has implications for the selective regulation of KATP channels across tissues. For example, tolbutamide has no effect on cardiac channels, and although glibenclamide will block both β cell and cardiac KATP channels, its effects are readily reversible in cardiac tissue, but not in insulin secreting cells.35 Such differences arise from the fact that even though SUR2A, the SUR component of the cardiac KATPchannel, is approximately 70% homologous to SUR1,36 the molecules are predominantly different towards the C-terminal region; that is, the part of SUR containing the SURB. In contrast to the effects of sulphonylureas on KATP channels, insulinotropic agents such as the imidazoline phentolamine close KATPchannels by a direct interaction with Kir6.2 rather than SUR1.37
SURs are also important in determining the pharmacological regulation of KATP channels by “potassium channel openers” such as diazoxide, which is used in the treatment of HI, and levcromakalim, pinacidil, and nicorandil. Most potassium channel openers will activate vascular and cardiac KATP channels38; however, only diazoxide4 and diazoxide derivatives39are effective against the β cell KATPchannels.40 As with the effects of sulphonylureas on SURs, the explanation for this appears to be the location of the potassium channel opener binding site. Using a series of chimaeric SUR protein derivatives of SUR1 and SUR2, Uhde and colleagues33 have shown that two sequences of SUR1 are crucial for determining potassium channel opener binding and KATP channel activation. These are located within the final series of TMDs at amino acids 1059–1087 (potassium channel opener I) and 1218–1320 (potassium channel opener II), which correspond to part of the linking sequence between TMDs 13–14 and TMD sequence 16–17 (fig 5A). These regions flank the putative SURB site, and are thought to form a binding pocket for diazoxide on the β cell KATP channel.33Although the stoichiometry for diazoxide has yet to be determined, based upon studies of the effects of potassium channel openers on SUR2,33 it seems likely that four molecules of the drug are required to initiate opening of the channels. No mutations in these sequences of amino acids have yet been reported in patients with PHHI (table 1), but clearly defects in this part of SUR1 could result in insensitivity to diazoxide in vivo.
Mutations found in patients with persistent hyperinsulinaemic hypoglycaemia of infancy (PHHI) within the SUR1 and KIR6.2 genes
Gene defects and HI
The gene encoding SUR1 comprises 39 exon boundaries and is clustered with the KIR6.2 gene—a single open reading frame lying immediately 3′ of the SUR1 gene and separated by only 4.5 kilobase pairs.52 Their location in the human genome on the short arm of chromosome 11 corresponds to a genetic locus linked to familial forms of a potentially lethal childhood disorder associated with persistent hypoglycaemia as a result of HI (B Glaseret al, 1999, unpublished).53The evidence is now convincing that gene defects within SUR1 or KIR6.2 can be correlated with the functional loss of KATP channels in β cells. However, this does not cause insulin hypersecretion per se; rather, loss of these channels leads to a dysregulation of Ca2+ homeostasis in β cells and unregulated insulin release is a consequence.
KATP channels and HI
Hypoglycaemia is a relatively common childhood metabolic abnormality and when persistent or recurrent is most frequently a consequence of hyperinsulinism: neonatal hyperinsulinism (OMIM:256450). Until recently, the pathophysiology of this group of conditions was not understood, although a defect in β cell function was first suggested in 1981, when it was shown that glucose failed to promote a concentration dependent release of insulin in tissue isolated from a child with hyperinsulinaemia induced hypoglycaemia.54
PHHI
PHHI is a severe form of HI, and a condition that is usually unresponsive to medical treatment and associated with profound hypoglycaemia in the early neonatal period. Sporadic cases are thought to be rare in the general population (approximately one in 45 000), but the disease has an incidence in communities with high rates of consanguinity approaching that of cystic fibrosis in white European populations—approximately one in 2500 live births.53Severe forms usually present within the first few hours or days of birth, as the result of extreme and sustained low blood glucose concentrations caused by unregulated secretion of insulin. Failure to recognise and promptly treat hypoglycaemia carries a substantial risk of severe brain damage and mental retardation because of a lack of fuels to sustain normal brain metabolism (for review see Glaser and colleagues53). Medical treatment for the disorder involves an increased carbohydrate intake to meet the raised requirement, and usually one or more drugs that inhibit insulin secretion. These agents include diazoxide, which was first introduced to treat hyperinsulinism in the 1960s, and somatostatin, both of which will activate KATP channels in normal β cells and inhibit insulin release.4 This occurs as a result of increased K+ channel opening causing a hyperpolarisation of the membrane and closure of voltage dependent Ca2+ channels. Unfortunately, however, the responsiveness of children with PHHI to these agents is highly variable, and patients who do not show an adequate and immediate response require surgery to remove their pancreas—usually a 95% pancreatectomy—to prevent recurrent hypoglycaemia. The variability in sensitivity to medical treatment is largely unexplained, but defects in the ionic control of insulin release prevail in PHHI β cells, and these can largely account for the findings.55-57 Pancreatectomy usually induces a clinical remission of symptoms, but this procedure will invariably predispose the patient with PHHI to pancreatic insufficiency and the development of diabetes mellitus in later life.
The correlation of KATP channel defects with PHHI
The genetic basis of PHHI has not been determined conclusively.53 In those cases where mutations have been linked to the disease,58 defects in the SUR1 and KIR6.2 genes are mainly, but not exclusively, inherited in an autosomally recessive manner (table 1).41 Gene defects in SUR1 can also be inherited differently. In these cases, the patient inherits a single paternal recessive SUR1 gene mutation, and a part of the pancreas is reduced to hemizygosity because of the loss of maternal imprinted genes.46 ,59 Loss of heterozygosity in the affected β cells results in insulin hypersecretion, but because of the loss of other maternally expressed imprinted genes (such as the tumour supressor genes H19 and p57KIP2), the pancreata of patients with this condition also have a morphologically distinctive appearance, with focal regions of β cell hyperproliferation. This has given rise to the term “focal PHHI” as distinct from “diffuse PHHI”, which is not associated with the loss of imprinted genes.47
For a number of SUR1 and KIR6.2 mutations that have been described it has now been shown that these gene defects can produce a variety of abnormalities in recombinant KATP channel function, including trafficking defects, assembly defects, and regulatory problems.58 However, very few studies have been undertaken in β cells isolated from patients with the disease. There are perhaps two reasons. First, it is technically difficult to isolate β cells from human tissue. Second, the amounts of tissue available for experimental studies after processing of the pancreas are extremely small. We have now undertaken studies on 49 patients with PHHI, and this work has been used to provide a crucial link between documenting gene variations in SUR1 and KIR6.2 and the clinical manifestation of hyperinsulinaemia induced hypoglycaemia.
Patch clamp studies (fig 6) require very limited amounts of primary tissue, and at the same time provide precise details about the molecular mechanisms of cellular control. Thus, using insulin secreting cells isolated after pancreatectomy, these experiments have shown that KATP channels are defective in PHHI β cells.42 ,48 ,55 ,56 ,60 The integration of PHHI β cell physiology with patient genotyping for KIR6.2 and SUR1 related defects is clearly an important consideration in these studies. In some studies, we have documented the complete absence of any operational KATP channels (R1437Q(23)X),48 whereas in another tissue, KATP channels were recorded, but they were devoid of regulation (exon 4, V187D mutation).42 It has not been possible to identify SUR1/KIR6.2 defects in all the patients studied, but the prevailing observations are that the loss of a crucial number of functional channels prevents the PHHI β cells from regulating the cell membrane potential.
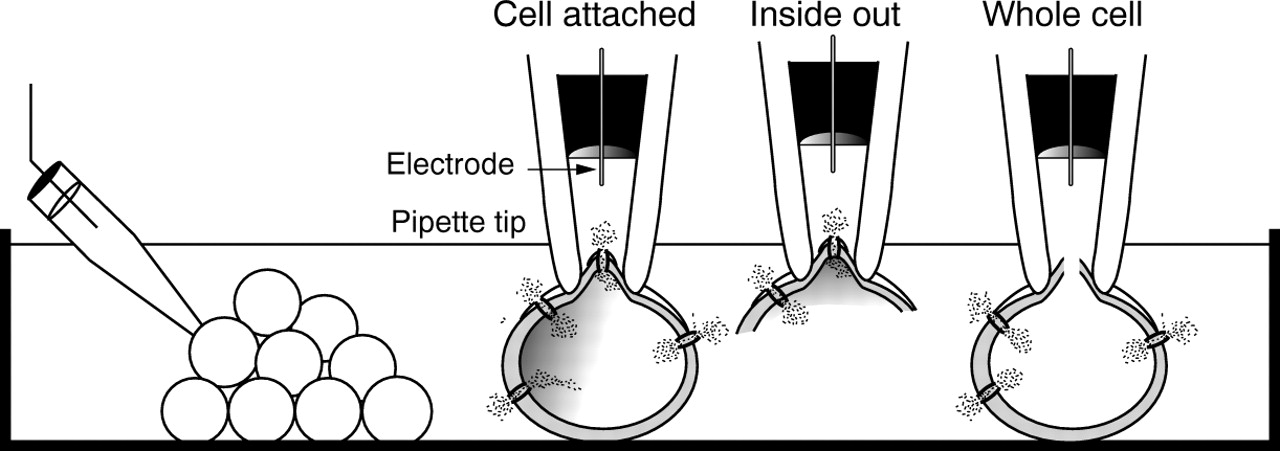
Patch clamp studies of ion channels in β cells. Patch clamp recordings are generated by “sealing” a glass micropipette (tip diameter ∼ 1 μm) against the outer membrane of a cell. Three of the various recording configurations are illustrated: the intact cell or “cell attached” patch, the cell free “inside out” patch, and the “whole cell” configuration. Note that single channel current events are recorded with the cell attached and inside out modes, whereas the whole cell configuration is used to record macroscopic currents.
In isolated PHHI β cells it has been found that the resting cell membrane potential is close to the threshold for the activation of voltage dependent Ca2+ channels, and it is activity from these channels that leads to the appearance of spontaneous, regenerating action potentials in the unstimulated PHHI β cell.56 Because Ca2+ influx is a key determinant of insulin secretion under normal conditions (fig 2), inappropriate Ca2+ channel activity will readily account for the unregulated secretion of insulin in PHHI (fig 1B).
Recordings of action potential currents from PHHI β cells indicate that they are regulated, as in normal β cells, by changes in the cell membrane potential. Thus, under voltage clamp conditions, hyperpolarisation of the membrane will terminate action potential firing and lead to the inhibition of voltage gated Ca2+channels. The consequences of this are important. First, the findings suggest that agents that hyperpolarise the β cell by non-KATP channel dependent events—for example, α2-agonists,61 might be clinically beneficial in alleviating hypoglycaemia (fig 7). Second, because a link between loss of KATP channels and insulin hypersecretion has been established, the implications are that blockers of voltage gated Ca2+ channels might be of value in the clinical management of PHHI.60

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Ion channels in insulin secreting cells. β Cells possess a number of ion channel families that are important in the control of the regulated release of insulin. In addition to the KATP channel, other important K+ channels include: the large conductance Ca2+ and voltage gated or KCa channel, the delayed rectifier K+ channel, a low conductance receptor mediated K+ channel (KI), and a K+ channel identified in persistent hyperinsulinaemic hypoglycaemia of infancy (PHHI) β cells as having sulphonylurea sensitivity and to be regulated by diazoxide, the KPHHI channel.55 The β cell possesses tetrodotoxin sensitive voltage gated Na+ channels, which contribute to the depolarisation phase of the action potential. The major Ca2+ channels in β cells are voltage activated and therefore responsible for Ca2+ entry after depolarisation of the membrane. In human β cells, two types have been described—the L-type (long lasting) and T-type (transient)—and there is also evidence for “non-T-type” and “N-type” channels. At least two types of Cl− selective anion channels are present: one of which is Ca2+ regulated whereas the other is cAMP regulated. Finally, two types of non-selective cation channels have been described, which are permeable to Na+, K+, and Ca2+, and might be involved with the refilling of intracellular stores after agonist induced stimulation: non-selective cation (ICRAN) channels.
Uncontrolled Ca2+ signals and treatment for HI
Nifedipine was first used in PHHI treatment after in vitro studies documented the appearance of spontaneous electrical activity in PHHI β cells and their inhibition by verapamil. Postoperatively, the patient continued to experience hypoglycaemic episodes and the nifedipine option was then introduced to prevent a second surgical resection of the pancreas. This resulted in both a stabilisation of blood glucose concentrations and an increased tolerance to fasting in the patient.60 Since then there have been other successful reports of nifedipine treatment,62 but nifedipine regimens can also be ineffective in the control of hypoglycaemia.63The lack of responsiveness of patients with HI to blockers of voltage gated Ca2+ channels is surprising, but recent findings might provide a clue to understanding this.
We investigated the mechanism of Ca2+ dependent insulin secretion using β cells isolated from 11 patients with PHHI after pancreatectomy. In these tissues we found great variability in the function of voltage gated Ca2+ channels. Of particular importance was the fact that it was not possible to record functionally operational channels in six of 11 patients, although they were recorded in 100% of normal human β cell controls.63 What is not yet clear is the mechanisms that determine an apparent causal relation between loss of KATP channels and defects in voltage gated Ca2+ channels. It is possible that the membrane environment of the PHHI β cell, which is permanently depolarised, leads to the downregulation of voltage gated Ca2+ channels, either at a functional level or by preventing gene transcription. Equally, it is also possible that SUR1 dependent interactions with voltage gated Ca2+ channels are involved. A key question that arises from this work is: how can insulin release be controlled if voltage gated Ca2+ channels fail to operate in PHHI β cells? One explanation for this is that other Ca2+ selective ion channels are active in PHHI β cells as a consequence of the primary defect in KATP channels. Evidence to support this possibility comes from in vitro studies using the agent maitotoxin—a dinoflagellate toxin and activator of ICRAN channels in β cells (figs 2 and 7). In PHHI β cells that lack voltage dependent mechanisms to increase the cytosolic Ca2+ concentration, we have shown that maitotoxin readily raises cytosolic Ca2+concentrations, thus implicating ICRAN channels in the pathogenesis of PHHI.63 Together, these findings could account for both the clinical variability of patients' responses to nifedipine, and unregulated insulin release in PHHI.
Other K+ channels and PHHI β cells
In β cells, several types of K+ channels are known to contribute to the complex electrophysiological profile that is generated in the presence of stimulatory concentrations of glucose (fig7). This essentially consists of cycles or waves of electrical activity with superimposed action potentials. Whereas closure of KATP channels crucially determines the initial depolarisation of the cell membrane, other K+ channels are subsequently involved in the generation of action potentials and establishing the oscillations of electrical activity.4 The delayed rectifier (KV) and Ca2+ and voltage gated (KCa) K+ channels are the major voltage dependent K+ channels in β cells, and contribute to the repolarisation phase of the action potential.64-68 In recent experiments, we made several conclusions about the properties of KV and KCa channels in β cells isolated from seven patients with PHHI.69 First, we found that the channels were functionally operational and that they were voltage dependent. Second, the voltage dependency of activation varied little between control (human) and disease tissue. Finally, we found that the numbers of channels in each cell were not significantly different between control and PHHI, and neither were there any major differences in their pharmacological properties. Only one minor aspect of the biophysical properties of KCa and KV channels was found to be different in PHHI β cells: an increase in the magnitude of currents at membrane potential values outside of the normal physiological range. This could have arisen from a shift in the voltage dependent activation profile of these channels, an effect that would make these channels relatively more sensitive to depolarisation of the membrane. Such findings are clinically relevant to PHHI treatment, because they suggest that selective activators of KCa and/or KV channels, were they available, might be potentially useful as alternatives to diazoxide and somatostatin at inhibiting insulin release in vivo.
Animal models of PHHI
Transgenic animal models of PHHI have also been developed in which the consequences of KATP channel dysfunction were studied in vivo. In one example of this work, transgenic mice were engineered to express a “dominant negative” version of Kir6.2 in β cells.70 Substitution of a glycine residue for serine at position 132 within the K+ ion conducting region of Kir6.2 terminates KATP channel activity, thereby mimicking this pathophysiological aspect of PHHI β cells. Affected mice were found to develop hyperinsulinaemic hypoglycaemia during the neonatal period, and β cells isolated from the animals had remarkably similar properties to human PHHI β cells: no KATP channel function, depolarised resting membrane potentials, and raised concentrations of cytosolic Ca2+. As the animals develop, hypoinsulinaemia and hyperglycaemia (that is, the onset of diabetes) follow the period of hyperinsulinaemic hypoglycaemia. These animals were found to have a higher frequency of apoptotic β cells in the prediabetic period compared with control animals, and for the first time this implicated programmed cell death as the reason for loss of β cells in PHHI.70 The concept of β cell “burn out” has a direct clinical correlation. In a recent review Glaser and colleagues53 discuss how long term, intensive treatment of patients with PHHI with somatostatin leads to remission of symptoms without the need for surgery. In such cases, it seems likely that this is caused by the long term destruction of β cells, which might result from unregulated Ca2+ entry, or indeed be a direct consequence of somatostatin treatment, which is known to be apoptogenic.71
KIR6.2 knockout (KIR6.2−/−) animals have also been created.72 In these animals, Kir6.2 function was specifically and selectively eliminated from pancreatic β cells. The developmental profile of these animals might have been expected to parallel that of the “dominant negative” mice; however, this was not the case. Quite surprisingly, β cells from KIR6.2−/− animals were less reminiscent of the human PHHI β cell than insulin secreting cells isolated from “dominant negative” mice.73 One other interesting feature of the KIR6.2−/− mice is that despite having impaired glucose dependent insulin release, both in vitro and in vivo, they have normal postprandial blood glucose values.72 This might implicate a negative correlation between Kir6.2 operation and insulin receptor expression in skeletal muscle tissue, which will tend to improve peripheral glucose clearance despite impaired insulin secretion. A SUR1 knockout (SUR1−/−)” mouse has also been developed recently.74 In these animals, the β cell resting membrane potential is depolarised with respect to normal values, and spontaneous Ca2+ action potentials are inhibitable by nifedipine. These properties are once again reminiscent of the human PHHI β cell; however, the physiological regulation of glucose homeostasis differs from the human disease, in that SUR1−/− animals apparently fail to increase insulin output in response to an increased glucose load.74
HI and acquired defects in KATP channel operation
Some children with congenital hyperinsulinism present with milder symptoms of hyperinsulinaemic induced hypoglycaemia than is described for PHHI, often with episodes of hypoglycaemia that are sporadic, occur postprandially, and are sensitive to diazoxide.75 ,76 In addition, some patients also present symptoms much later in life, even in adulthood.77 In these cases, hyperinsulinism can be inherited in an autosomally dominant fashion and is not principally linked to defects in the SUR1 or KIR6.2 genes. So far, mutations in two other genes, both associated with glucose homeostasis and acquired KATP channel dysfunction in β cells, have now been shown to give rise to clinically distinct forms of HI: HI-GK and HI-GLUD1 (fig 1C).
HI-GK arises from defects in the glucokinase gene (GK).75Gene mutations that decrease the sensitivity of the enzyme for glucose are known to give rise to maturity onset diabetes of the young type 2.78 In contrast, the mutations described in patients with HI-GK result in the generation of an “activated” gene product with a greatly increased sensitivity to glucose. It has been suggested that these mutations result in excessive ATP production in β cells and the inappropriate closure of KATP channels (fig 1C).
The GLUD1 gene encodes the enzyme glutamate dehydrogenase (GDH), which links glutamate metabolism with the citric acid cycle and catalyses the conversion of glutamate to α-ketoglutarate. The term HI-GLUD1 has been introduced to describe patients who present with a clinical phenotype of hyperinsulinism associated with hyperammoneamia.76 Under normal conditions, the activity of GDH is controlled by allosteric inhibition by GTP. In some patients with HI-GLUD1, defects in the region of GLUD1 encoding the GTP binding domain of the enzyme have been identified that lead to a decrease in the sensitivity of GDH to GTP. This results in an “activated enzyme” complex, which in the liver results in excessive ammonia production, whereas in β cells, after dietary intake, leucine induced stimulation of glutamate dehydrogenase is presumed to increase the flux of glutamine into the citric acid cycle. As in the case of HI-GK, the predicted β cell response is a net increase in the cytosolic ATP concentration, leading to enforced closure of KATPchannels, a constant depolarisation of the membrane, uncontrolled voltage gated Ca2+ channel activity, and hyperinsulinism as a consequence (fig 1C). Recently, a different mode of GDH activation was described in a patient with severe hyperinsulism hyperammonemia syndrome.79 In this case, the patient was heterozygous for two mutations outside of the GTP binding domain of the enzyme (A1401C, N410T), which were associated with a constitutively high level of GDH activity.
Therefore, the loss of KATP channel operation is a common pathophysiological event in the onset of HI-GK, HI-GLUD, and PHHI. However, it should also be noted that because KATP channels are “operational” in patients with HI-GK and HI-GLUD, these patients show good clinical responses to diazoxide, whereas patients with PHHI do not.
NES2Y: a PHHI derived human β cell line
While studying the physiology of PHHI β cells we discovered recently that these cells have the potential to proliferate spontaneously in vitro. We have studied one β cell line—designated NES2Y—in detail.80 NES2Y cells reproduce the properties and key features of acutely isolated PHHI β cells, namely: (1) they lack operational KATP channels; (2) they have greatly impaired cytosolic Ca2+ signalling mechanisms; (3) they constitutively release insulin at an increased rate in the absence of stimuli; and (4) they show a limited responsiveness to glucose through the KATP channel independent or glucose augmentation pathway.9 Because PHHI is a rare condition, the availability of these cells is an important asset to ongoing studies of the molecular pathophysiology of the condition.
In vitro gene therapy for PHHI
In addition to loss of KATP channel operation, NES2Y β cells also have impaired expression of the homeodomain transcription factor PDX1.18 In recent studies, we found that defects in the functional operation of NES2Y β cells can be overcome by triple transfection of the cells with cDNA molecules encoding SUR1, Kir6.2, and PDX1. This has led to the generation of a fully glucose responsive human β cell line—designated NISK9 cells.80 The properties of KATP channels expressed in the NISK9 β cells are identical to those in native tissue (inwardly rectifying, inhibited by cytosolic ATP in a concentration dependent manner, activated by ADP in the presence of ATP, modulated by diazoxide and tolbutamide, and so on), and their operation has clear functional consequences. Not only did the transfection event underpin the development of glucose responsiveness within a physiologically relevant concentration range, but it also led to a considerable decrease in the rate of insulin release under basal conditions. This has provided further proof of the causal relation between KATP channel defects and insulin hypersecretion in PHHI.
It is currently unclear why NES2Y cells (and other PHHI derived β cells) proliferate in culture. Because these cells have defects in PDX1 expression,18 and because PDX1 is involved in pancreatic β cell lineage determination,17 NES2Y cells may represent a stage in islet cell development at which the cells have retained the ability to replicate while attaining a β cell like phenotype. The wider implications of this possibility and the role of PDX1 in HI clearly need to be investigated. Alternatively, it is also possible that unregulated Ca2+ influx in PHHI derived tissue is having a permissive role on Ca2+ dependent genes associated with cell cycle events.
Concluding remarks
Since the genes associated with HI were first identified in 1994–5, we have moved in a relatively short period of time to a position of understanding the defective gene products, and how these impact upon the physiology of insulin release. A meaningful interface of applied physiology and clinical medicine has been established, and it is already clear that information gained from pathophysiological studies is now being used in the clinical environment, both in terms of prognosis and management of the condition (A Aynsley-Greenet al, 1999, unpublished).81Armed with a greater awareness of the pathophysiology of PHHI β cells, and the availability of an in vitro model system, it might also be possible in the future to isolate β cells from patients with PHHI after pancreatectomy, and genetically engineer the cells for subsequent autotransplantation. Clearly, this is a long term objective, but one that can now be advanced through the availability of PHHI derived β cell lines.
Acknowledgments
Work in our laboratory is supported by the British Diabetic Association and the Medical Research Council. This collaborative group initiative was supported by a European Union funded concerted action grant (BMH4-CT98–3284) from the European Network for Research into Hyperinsulinism in Infancy (ENRHI).