
Abstract
Numerous recent reports have proposed that mutations in the C-terminal domain of the MECP2 gene could be a frequent cause of mental retardation in males. We have identified two mutations in this particular domain (S359P and E397K) in two boys who were screened for MECP2 mutations in a series of 23 mentally handicapped boys fitting the clinical description of the previously reported cases. A detailed familial study based on three generations shows that the first mutation (S359P) was also inherited by a healthy cousin thus ruling out its involvement in the etiology of the phenotype of this patient. The second mutation (E397K) was also found in normal individuals. These findings clearly call for a careful consideration of the pathogenicity of the MECP2 mutations identified in sporadic male cases before genetic counselling or prenatal diagnosis is proposed to the corresponding families.
Similar content being viewed by others

Introduction
Mutations in the methyl-CpG binding protein 2 (MECP2) gene located in Xq28 are the cause of Rett Syndrome, a severe neurodevelopemental disorder affecting only females.1 Mutations in this gene were also found in mentally retarded males with a wide range of phenotypes varying from lethal neonatal encephalopathy to moderate mental retardation2,3,4,5,6,7,8,9,10 In a recent study, the frequency of MECP2 mutations was estimated to be around 2% in a large series of mentally retarded males.7 These findings suggest that MECP2 should be analysed extensively in mentally retarded males and could therefore result in significant progress in elucidating the etiology of mental handicap.
The MECP2 gene is composed of four exons with the coding sequence in exons 2–4. The 3′-untranslated region (3′-UTR) of exon 4 is unusually long and well-conserved between human and mouse, although its functional significance has not been elucidated. This gene encodes a 486 amino-acid methyl-CpG binding protein involved in gene silencing through a mechanism that implicates binding to mCpG and recruitment of histones deacetylases.11,12,13 Defects in MECP2 function are expected to lead to the abnormal transcription of a number of currently unidentified genes.
In order to further analyse the role of MECP2 mutations in the etiology of male mental retardation, 23 unrelated patients were screened for mutations in this gene. Inclusion criteria were severe mental retardation either with a clinical history and phenotype reminiscent of male Rett syndrome variant as described previously,14 or patients with Angelman-like phenotype. In this latter case, diagnosis of Angelman syndrome was excluded after clinical examination and molecular analysis of the methylation pattern of the 15q11-q12 region (data not shown). To screen for mutations, we performed direct sequencing of the three coding exons of the MECP2 gene for each patient.
Materials and methods
MECP2 mutation screening
The three coding exons were amplified using PCR and genomic DNA as a template. Both strands were sequenced. Sequencing reactions were run on a LiCor automated sequencer.
X-chromosome inactivation analysis
Primers were designed in the (CAG)n flanking sequences within the first intron of the Androgen receptor (HUMARA) gene. The forward primer AR-P1 (5′ TCC AGA ATC TGT TCC AGA GCG TGC 3′) was 5′ labelled (IRD800) and the reverse primer AR-P2 (5′ GCT GTG AAG GTT GCT GTT CCT CAT 3′) was unlabelled. Four hundred nanograms of DNA were digested by HpaII and ethanol precipitated. One hundred ng of both HpaII digested DNA and total uncut DNA from each individual was used as a template for PCR. One-fiftieth of the PCR product was diluted in loading buffer and directly loaded onto a LiCor automated sequencer. Analysis of the relative amount of each allele was done using the One-D-Scan software (Scanalytics).
Results
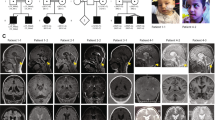
We have identified a T to C transition in exon 3 of MECP2 resulting in a S359P amino-acid substitution in one patient. This patient was referred for suspicion of Angelman syndrome at 18 months of age (Figure 1). He was the first child of healthy parents with an unremarkable familial history. At birth, he was an apparently normal boy. After surgical treatment of a pyelo-ureteric junction obstruction at 2 months of age, a regression of the psychomotor development was observed associated with abnormal movements of the mouth and tongue. Sitting unaided was acquired at 13 months and walking was not achieved at 18 months of age. Neurological examination showed spasticity with ataxia. He had no speech development. He presented a poor eye contact, frequent facial grimacing, episodic inappropriate laughter or unexplained crying, often nocturnal, repetitive hand stereotypies such as tapping, hand bitting and washing as well as hyperventilation and teeth grinding. Physical examination showed a postnatal microcephaly (-2SD) and a mild facial dysmorphism with deep set eyes, malar hypoplasia, thin upper lip and large ears. Complex partial seizures occurred at this age. Electroencephalogram showed slow background activity with paroxysmal slow spike waves. Brain MRI was unremarkable. RHG-banding standard karyotype was normal. At 9 years of age, he was a severely mentally handicapped boy. Walking was acquired at 8 years, but remained unstable with a wide base and ataxic gait. Distal lower limb muscles atrophy and peripheral vasomotor disturbances were noticed. Frequent episodes of hypothermia and chronic constipation were reported by the parents. His behaviour remained unchanged. However, he had better eye contact and was able to use a few signs to communicate.

Picture of the patient carrying the S359P mutation.
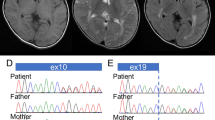
To determine if the missense mutation found in the C-terminal domain of MECP2 was the cause of mental retardation in this patient, his relatives were analysed (Figure 2). The healthy brother of the patient did not carry the S359P mutation but their mother was a carrier. We performed X-chromosome inactivation (XCI) analysis on the mother's DNA samples extracted from fresh blood, hair root and buccal mucosa, to determine if she displayed a favourably skewed XCI pattern. This analysis was performed at the androgen receptor locus (AR, Xq12) and the relative amount of each allele was determined on undigested or HpaII digested DNA as described elsewhere.6 The carrier mother displayed a random XCI pattern in all the tissues examined (67 : 33, 41 : 59 and 55 : 45 for blood, hair root and buccal mucosa respectively). Since these results were not satisfactory to provide clear genetic counselling to the family, we next screened further relatives for the same mutation. This analysis showed that the mother's sister and her three healthy children were also carriers of the S359P mutation (Figure 3), thus ruling out its involvement in the etiology of the phenotype of the proband.

Pedigree of the family with the S359P mutation. The proband is indicated by an arrow. Carriers of the S359P mutation are indicated by a black dot into their respective symbols. Individuals II-1 and III-5 do not carry the S359P mutation.

Determination of the carrier status in the family with the S359P mutation. In each case, a 610 bp PCR fragment spanning the mutation was amplified using genomic DNA as a template. This product contains two BseRI sites: one at 175 bp and one at 596 bp. The mutation suppresses the BseRI restriction site located at 175 bp. The purified PCR product was digested (+BseRI lanes) or undigested (−BseRI lanes) to detect the mutation. A wild-type allele produces three restriction fragments of 14, 175 and 421 bp (the 14 bp fragment is not visible on this gel). A mutated allele produces two restriction fragments of 596 and 14 bp. Individuals II-2, III-1, III-2 and III-3 are carriers. 1347 is a control female DNA. Each fragment was also sequenced to confirm the presence of the mutation by an alternative technique (data not shown).
We have also found in another mentally retarded male of our series the E397K mutation which was described previously as a polymorphism.2 No further analysis was performed in this case.
Discussion
Up to now, a total of 14 MECP2 mutations have been reported in male cases (Table 1). The first two cases reported in the literature2,6 demonstrated the existence of a variant phenotype of Rett syndrome in males characterised by a severe neonatal encephalopathy and lethality in early childhood due to breathing failure. In both cases, the carrier mothers had a totally skewed X-chromosome inactivation. Next, it was shown that MECP2 mutations can be compatible with life after childhood in male cases.5 Further mutations were found after screening of families with non-specific X linked mental retardation previously linked to Xq28, but also sporadic male cases with a wide spectrum of mental retardation varying from a severe form of mental retardation like Angelman-like patients but also moderately mentally retarded males. A recent report states that mutations in the C-terminal domain of the MECP2 gene are a frequent cause of mental handicap.7 Three missense mutation, G428S, R453Q and P399L have been found associated with an heterogeneous phenotypic spectrum varying from severe to moderate mental retardation and are reported as disease-causing.7,9 It is interesting to observe that the X-chromosome inactivation pattern of the carrier mothers was found to be random when tested, contrasting with the non-random XCI pattern in carriers of MECP2 mutations associated with fatal neonatal encephalopathy in males.
The S359P mutation reported here was found both in a patient fulfilling the criteria of male variant Rett syndrome and a healthy cousin, hence demonstrating that it is a benign variant. The same conclusions apply for the E397K mutation, which was also found in normal individuals. Both amino-acids are conserved in the mouse and rat Mecp2 proteins but not in that of Xenopus laevis (Table 1). However, since C-terminal mutations do not fall within a known functional domain of the MECP2 protein, it is difficult to draw conclusions about the significance of these observations.
Our findings highlight the need for extreme caution in the clinical interpretation of sequence variation in the MECP2 gene, especially when these are missense mutations and when they fall outside a well-characterised functional domain. The genetic counselling issue is of particular importance since most of the mothers of such male patients are carriers of the mutation, in contrast to what is found in Rett syndrome girls. In the two cases reported here, the mutation is not the cause of the phenotype. In the light of these observations, it is thus highly important to carefully consider the arguments favouring pathogenicity of the MECP2 mutations identified in sporadic male cases before genetic counselling or prenatal diagnosis is proposed to the corresponding families.
References
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi H . Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2 Nat Genet 1999 23: 185–188
Wan M, Lee SSJ, Zhang X et al. Rett syndrome and beyond: recurrent spontaneous and familial MECP2 mutations at CpG hotspots Am J Hum Genet 1999 65: 1520–1529
Clayton-Smith J, Watson P, Ramsden S, Black GC . Somatic mutation in MECP2 as a non-fatal neurodevelopmental disorder in males Lancet 2000 356: 830–832
Meloni I, Bruttini M, Longo I et al. A mutation in the Rett syndrome gene, MECP2, causes X-linked mental retardation and progressive spasticity in males Am J Hum Genet 2000 67: 982–985
Orrico A, Lam C, Galli L et al. MECP2 mutation in male patients with non-specific X-linked mental retardation FEBS Lett 2000 481: 285–288
Villard L, Kpebe A, Cardoso C, Chelly PJ, Tardieu PM, Fontes M . Two affected boys in a Rett syndrome family: clinical and molecular findings Neurology 2000 55: 1188–1193
Couvert P, Bienvenu T, Aquaviva C et al. MECP2 is highly mutated in X-linked mental retardation Hum Mol Genet 2001 10: 941–946
Hoffbuhr K, Devaney JM, LaFleur B et al. MeCP2 mutations in children with and without the phenotype of Rett syndrome Neurology 2001 56: 1486–1495
Imessaoudene B, Bonnefont JP, Royer G et al. MECP2 mutation in non-fatal, non-progressive encephalopathy in a male J Med Genet 2001 38: 171–174
Leonard H, Silberstein J, Falk R et al. Occurrence of Rett syndrome in boys J Child Neurol 2001 16: 333–338
Lewis JD, Meehan RR, Henzel WJ et al. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA Cell 1992 69: 905–914
Nan X, Ng HH, Johnson CA et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex Nature 1998 393: 386–389
Chandler SP, Guschin D, Landsberger N, Wolffe AP . The methyl-CpG binding transcriptional repressor MeCP2 stably associates with nucleosomal DNA Biochemistry 1999 38: 7008–7018
Jan MM, Dooley JM, Gordon KE . Male Rett syndrome variant: application of diagnostic criteria Pediatr Neurol 1999 20: 238–240
Acknowledgements
We thank the families for their cooperation and the referring clinicians. We thank Pr Nicole Philip for clinical expertise and helpful comments.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Moncla, A., Kpebe, A., Missirian, C. et al. Polymorphisms in the C-terminal domain of MECP2 in mentally handicapped boys: implications for genetic counselling. Eur J Hum Genet 10, 86–89 (2002). https://doi.org/10.1038/sj.ejhg.5200761
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5200761
